锁阳及其制剂特征图谱的构建方法及原儿茶酸含量的检测方法与流程
1.本发明属于药物检测技术领域,具体涉及一种锁阳及其制剂特征图谱的构建方法及原儿茶酸含量的检测方法。
背景技术:
2.中药锁阳为锁阳科(cynomoriaceae)锁阳属(cynomorium)植物锁阳(cynomorium songaricum-rupr.)的干燥肉质茎,又名铁棒槌、乌兰高腰(蒙语)。锁阳性味甘、温,归肝、肾、大肠经,始载于《本草衍义补遗》及《本草纲目》,具补肾阳、益精血、润肠通便之功效,主要用于肾阳不足、腰膝酸软、精血亏虚、阳痿、滑精、肠燥便秘等疾病的治疗。民间素有“不老药”的美誉。主要分布于内蒙古、宁夏、新疆、甘肃、青海等西北地区,是中药和蒙药中的常用药物。
3.现代药理学研究表明,锁阳具有滋补强壮、增强免疫、抗衰老、抗应激、清除自由基、抑制血小板凝集等作用。锁阳中的有效成分有以儿茶素为代表的黄酮类、原儿茶酸为代表的有机酸类、熊果酸为代表的三萜类、多糖与鞣质等化合物为主。原儿茶酸具有与锁阳相近的药理作用,能够发挥增加冠脉流量、降低心肌耗氧量、抑制血小板聚集、抗菌、祛痰、平喘和解蛇毒等作用。
4.目前,市售的锁阳药材普遍存在掺杂、混用、乱用等现象。《中国药典》2015版一部只收载了以脯氨酸和熊果酸为对照品的薄层鉴别锁阳的方法,没有具体的含量测定指标和指纹图谱检测方法,难以全面反映锁阳内在质量。
5.近年来,分析测试工作者们在锁阳的质量控制方面做了很多工作,例如,王勤等采用高效液相色谱法测定不同地区、不同生育期锁阳的原儿茶酸含量的动态变化,该方法供试品溶液的制备方法复杂,多次萃取易造成供试品溶液间的不平行,耗时费力。刘晔伟等利用hplc建立锁阳指纹图谱,提出同时测定儿茶素和根皮苷的方法,但其分析时间较长,图谱中干扰峰较多,此外,对于锁阳中易溶于水的没食子酸、原儿茶酸等关键成分均未做指认。
6.中国专利文献cn107290442a,公开了一种使用高效液相色谱法同时测定锁阳样品中没食子酸、原儿茶酸、儿茶素及根皮苷四种化学成分的方法,但是该方法分析时间较长,供试品溶液制备过程复杂,得到的特征图谱中的色谱峰会出现堆积的问题。
技术实现要素:
7.因此,本发明要解决的技术问题在于克服现有技术中锁阳液相色谱的表征方法中没有对关键成分有机酸进行指认,且色谱峰的分离效果差等缺陷,从而提供一种锁阳及其制剂特征图谱的构建方法及原儿茶酸含量的测定方法。
8.为此,本发明提供了以下技术方案。
9.本发明提供了一种锁阳及制剂特征图谱的构建方法,以原儿茶酸作为对照品,采用高效液相色谱法获得所述锁阳及制剂的特征图谱;
10.所述特征图谱至少包括4个特征峰,分别是1号峰、2号峰、3号峰和4号峰;以4号峰原儿茶酸为参照峰,其它特征峰的相对保留时间在规定值的
±
10%内,相对保留时间的规定值分别是:
11.1号峰的相对保留时间为0.47;2号峰的相对保留时间为0.54;3号峰的相对保留时间为0.81。
12.所述构建方法,包括以下步骤,
13.对照品溶液的制备:分别取原儿茶酸、没食子酸和儿茶素对照品,加入有机溶剂后制成对照品溶液;
14.供试品溶液的制备:取锁阳待测样品,加入醇溶液,提取,分离,取上清液,过滤后,即得;
15.色谱条件与系统适用性试验:按照高效液相色谱法测定,采用十八烷基键合硅胶或实心核颗粒为填充剂的色谱柱;以甲醇为流动相a,以甲酸溶液为流动相b,按照梯度洗脱程序进行色谱分析,流动相流速为0.1-0.5ml/min,检测波长为255-300nm;
16.测定:分别吸取对照品溶液、供试品溶液注入超高效液相色谱仪,测定,即得;
17.所述梯度洗脱的程序包括:0-9min,流动相a:流动相b为0%:100%。
18.所述梯度洗脱程序包括:0-9min,流动相a:流动相b为0%:100%;9-10min,流动相a:流动相b为0-10%:100-90%;10-13min,流动相a:流动相b为10%:90%;13-20min,流动相a:流动相b为10-14%:90-86%;20-26min,流动相a:流动相b为14-16%:86-84%;26-30min,流动相a:流动相b为16-18%:84-82%;
19.所述色谱条件:按照高效液相色谱法测定,采用waterst3色谱柱;以甲醇为流动相a,以体积分数0.2%甲酸溶液为流动相b,按照梯度洗脱程序进行色谱分析,流动相流速为0.3ml/min,检测波长为290nm。
20.所述特征图谱还包括5号峰、6号峰、7号峰和8号峰,以4号峰为参照峰,其它特征峰的相对保留时间在规定值的
±
10%内,相对保留时间的规定值分别是:
21.5号峰的相对保留时间为2.39;6号峰的相对保留时间为2.82;7号峰的相对保留时间为3.35;8号峰的相对保留时间为3.52。
22.进一步地,在所述供试品溶液的制备中,所述醇溶液是体积分数为20-50%甲醇溶液;
23.所述供试品溶液的浓度为0.1-0.3g/ml;在所述供试品溶液的制备中,所述提取方法为超声提取法或回流提取法;所述提取时间为15-25min;
24.在所述对照品溶液的制备中,所述有机溶剂为甲醇溶液;
25.所述对照品溶液的浓度为0.03-0.05mg/ml。
26.所述锁阳待测样品为锁阳药材、锁阳饮片、锁阳标准汤剂冻干粉或锁阳配方颗粒。
27.本发明还提供了一种锁阳中原儿茶酸含量的检测方法,包括以下步骤,
28.对照品溶液:取原儿茶酸对照品,加入有机溶剂后制成对照品溶液;
29.供试品溶液:取锁阳待测样品,加入醇溶液,提取,分离,取上清液,过滤后,即得;
30.含量测定:通过高效液相色谱法,测定对照品溶液和供试品溶液中原儿茶酸的峰面积,根据外标法计算锁阳中原儿茶酸的含量。
31.所述高效液相色谱法的色谱条件:按照高效液相色谱法测定,采用十八烷基键合
硅胶或实心核颗粒为填充剂的色谱柱;以甲醇为流动相a,以甲酸为流动相b,按照梯度洗脱程序进行色谱分析,流动相流速为0.1-0.5ml/min,检测波长为255-300nm。
32.所述梯度洗脱程序包括:0-9min,流动相a:流动相b为0%:100%;9-10min,流动相a:流动相b为0-10%:100-90%;10-13min,流动相a:流动相b为10%:90%;13-20min,流动相a:流动相b为10-14%:90-86%;20-26min,流动相a:流动相b为14-16%:86-84%;26-30min,流动相a:流动相b为16-18%:84-82%;
33.所述高效液相色谱法的色谱条件为,按照高效液相色谱法测定,采用waterst3为填充剂的色谱柱;以甲醇为流动相a,以甲酸为流动相b,按照梯度洗脱程序进行色谱分析,流动相流速为0.1-0.5ml/min,检测波长为260nm。
34.所述对照品溶液的制备步骤包括,取原儿茶酸对照品,加入甲醇后制成对照品溶液;
35.所述供试品溶液的制备步骤包括,取锁阳待测样品,加入甲醇溶液,超声提取或回流提取后冷却,分离,取上清液,过滤后,即得。
36.本发明技术方案,具有如下优点:
37.1.本发明提供的锁阳及其制剂特征图谱的构建方法,该方法重复性好、稳定性好、耐用性强;本发明采用高效液相色谱法,通过控制洗脱梯度程序和流动相系统,能够使锁阳特征图谱中的特征峰的峰形和分离度较好,同时还能以原儿茶酸、没食子酸和儿茶素等锁阳关键成分作为特征峰,为锁阳的质量控制提供了更为科学、合理、详实的依据,有利于分析锁阳品种差异和生长过程中各成分的积累变化规律。
38.该特征图谱包括至少4个特征峰,以原儿茶酸为参照峰,其它特征峰的相对保留时间在规定值的
±
10%内,相对保留时间的规定值分别是:1号峰的相对保留时间为0.47;2号峰的相对保留时间为0.54;3号峰的相对保留时间为0.81,该方法可以分离得到多个稳定、重复的色谱峰,准确度好,能够实现对锁阳及制剂的质量控制。
39.本发明提供的构建方法,可以在9min内,得到至少4个对锁阳及制剂质量控制的特征峰,检测效率高。
40.本发明提供的方法适用于锁阳药材、锁阳饮片、锁阳标准汤剂冻干粉或锁阳配方颗粒等特征图谱的构建。
41.2.本发明提供的锁阳及其制剂特征图谱的构建方法,该方法中供试品溶液的制备方法简单易重现,有助于锁阳中的有效成分被提取得到,为图谱提供更多的特征峰。
42.当检测波长为290nm时,特征图谱的信息最为丰富,检测效果最准确。
43.3.本发明提供的锁阳中原儿茶酸含量的检测方法,该方法采用高效液相色谱法,减少了对锁阳化学成分测定方面的资源和时间的浪费,能够全面、快速、准确的评估锁阳样品的质量。当检测波长为260nm时,原儿茶酸有最大的吸收,检测效果最准确。
附图说明
44.为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
748100-02;
70.批次3:190915-750306-03饮片产地:内蒙古阿拉善旗,饮片批号为yp190915-750306-03;
71.批次4:190915-750306-04饮片产地:内蒙古阿拉善旗,饮片批号为yp190915-750306-04;
72.批次5:190915-750306-05饮片产地:内蒙古阿拉善旗,饮片批号为yp190915-750306-05;
73.批次6:190915-750306-06饮片产地:内蒙古阿拉善旗,饮片批号为yp190915-750306-06;
74.批次7:190915-010000-07饮片产地:内蒙古左旗,饮片批号为yp190915-010000-07;
75.批次8:190915-010000-08饮片产地:内蒙古左旗,饮片批号为yp190915-010000-08;
76.批次9:190915-010000-09饮片产地:内蒙古左旗,饮片批号为yp190915-010000-09;
77.批次10:191023-025250-10饮片产地:内蒙古赤峰市林西县,饮片批号为yp191023-025250-10。
78.锁阳配方颗粒的制备方法包括:取锁阳饮片1900g,加水煎煮,滤过,浓缩,干燥,加入相当于饮片量13%的糊精,混匀,制粒,制成1000g,分装,即得。批号为kl190801-748100-02。
79.本发明用到的锁阳药材的批号为:yc190801-748100-02。
80.试剂:
81.甲醇(默克股份两合公司),色谱纯;
82.甲酸(国药集团化学试剂有限公司),分析纯;
83.磷酸(赛默飞世尔科技有限公司),色谱纯;
84.乙酸(国药集团化学试剂有限公司),分析纯;
85.乙腈(默克股份两合公司),色谱纯;
86.水为蒸馏水(屈臣氏)。
87.仪器:
88.waters acquityh-class超高效液相色谱仪,tuv detector检测器,empower 3色谱工作站;
89.ml204t电子天平(梅特勒
·
托利多)、xy2000-2c电子天平(常州市幸运电子设备有限公司)和msa6.6s-0ce-dm电子天平(赛多利斯科技仪器(北京)有限公司);
90.kq-100db超声波清洗器(昆山市超声仪器有限公司);
91.tg-16s高速离心机(四川蜀科仪器有限公司)。
92.实施例1
93.本实施例提供了一种锁阳及其制剂特征图谱的构建方法,包括以下步骤,
94.对照品溶液的制备:取3.999mg的原儿茶酸对照品,加入体积分数50%甲醇水溶液,得到0.04mg/ml原儿茶酸对照品溶液;
95.取4.012mg的没食子酸对照品,加入体积分数50%甲醇水溶液,得到0.04mg/ml没食子酸对照品溶液;
96.取4.001mg的儿茶素对照品,加入体积分数50%甲醇水溶液,得到0.04mg/ml儿茶素对照品溶液。
97.供试品溶液的制备:取锁阳待测样品1.0g,精密称定,置于具塞锥形瓶中,精密加入体积分数30%甲醇10ml,密塞,称定重量,超声(功率500w,频率40kh)处理20min,放至室温,补足减失的重量,摇匀,离心5min,转速为3500转,然后取上清液,过0.22μm微孔滤膜,得到1g/ml的供试品溶液。
98.色谱条件与系统性适用性试验:按照高效液相色谱法测定,采用waters t3(2.1
×
100mm,1.6μm)色谱柱,以甲醇为流动相a,以体积分数0.2%甲酸溶液为流动相b,按照梯度洗脱程序进行色谱分析,梯度洗脱程序为:0-9min,流动相a:流动相b为0%:100%;9-10min,流动相a:流动相b为0-10%:100-90%;10-13min,流动相a:流动相b为10%:90%;13-20min,流动相a:流动相b为10-14%:90-86%;20-26min,流动相a:流动相b为14-16%:86-84%;26-30min,流动相a:流动相b为16-18%:84-82%,流动相流速为0.3ml/min,检测波长为290nm;柱温35℃。
99.测定:分别吸取2μl对照品溶液和2μl供试品溶液注入超高效液相色谱仪,测定,得到特征图谱。
100.特征峰指认:
101.取10个批次的锁阳标准汤剂冻干粉待测样品,按照上述方法制成10批供试品溶液,吸取2μl注入超高效液相色谱仪,按照实施例1中的色谱条件,测定,得到10个批次锁阳待测样品的特征图谱。采用“中药色谱特征图谱相似度评价系统(2012版)”软件进行分析,时间窗设为0.1采用平均数法,以4号峰为参照峰,经多点矫正后自动匹配得到10批待测样品叠加图谱及其对照特征图谱,10个批次的供试品溶液的叠加色谱图见图1,生成的对照特征图谱参照图2。相似度评价结果见表1。
102.锁阳的特征图谱包括8个共有特征峰,分别是1号共有特征峰,2号共有特征峰,3号共有特征峰,4号共有特征峰,5号共有特征峰,6号共有特征峰,7号共有特征峰,8号共有特征峰;通过原儿茶酸对照品溶液、没食子酸对照品溶液和儿茶素对照品溶液的色谱图进行指认,原儿茶酸对照品的色谱图见图3,没食子酸对照品的色谱图见图4,儿茶素对照品的色谱图见图5,1号峰为没食子酸峰,4号峰为原儿茶酸峰,5号峰为儿茶素峰;以4号峰为参照峰(s峰),计算其它特征峰的相对保留时间,允许误差在规定值的
±
10%以内,相对保留时间的规定值分别是:
103.1号峰的相对保留时间为0.47;2号峰的相对保留时间为0.54;3号峰的相对保留时间为0.81;5号峰的相对保留时间为2.39;6号峰的相对保留时间为2.82;7号峰的相对保留时间为3.35;8号峰的相对保留时间为3.52。
104.表1 10批锁阳标准汤剂冻干粉的特征图谱相似度评价结果
105.序号批号与对照特征图谱相似度1190801-015000-010.9892190801-748100-020.9983190915-750306-030.972
4190915-750306-040.9655190915-750306-050.9906190915-750306-060.9837190915-010000-070.9988190915-010000-080.9939190915-010000-090.97710191023-025250-100.908对照图谱r1.000
106.表1中,10个批锁阳供试品溶液的相似度值在0.908-0.998范围之间,说明特征图谱之间的差异较小。
107.实验例1方法学考察
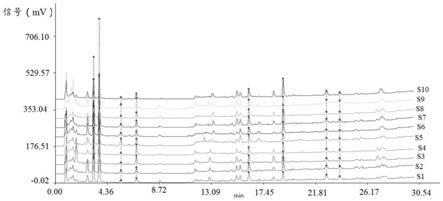
108.(1)溶剂的影响
109.取2μl体积分数30%甲醇水溶液注入超高效液相色谱仪,按照实施例1中的色谱条件,测定,得到的色谱图中无色谱峰,说明溶剂对锁阳特征图谱的色谱峰没有影响,色谱图见图6。
110.(2)稳定性试验
111.取同一批次的锁阳待测样品(批号190801-748100-02),按照实施例1中的方法制成供试品溶液,分别于室温条件下放置0h、2h、4h、6h、8h、10h、12h、24h后,按照实施例1中的色谱条件,测定,得到色谱图,以4号峰为参照峰,计算色谱图中共有峰的相对保留时间和相对峰面积,见表2和表3,结果显示,共有峰的相对保留时间rsd均小于1.0%,12h内相对峰面积的rsd均小于5.3%,表明样品在12h内稳定。
112.表2 特征峰的相对保留时间
[0113][0114]
表3 特征峰的相对峰面积
[0115][0116][0117]
(3)重复性实验
[0118]
取同一批次锁阳待测样品(批号190801-748100-02),按照实施例1中的方法制成6份供试品溶液,分别编号1-6,按照实施例1中的色谱条件,测定。结果,以4号峰为参照峰,计算色谱图中共有峰的相对保留时间和相对峰面积,见表4和表5,计算结果显示,共有峰的相对保留时间rsd均小于1.0%,相对峰面积的rsd均小于5.8%,表明该方法的重复性良好。
[0119]
表4 特征峰的相对保留时间
[0120][0121]
表5 特征峰的相对峰面积
[0122]
[0123][0124]
(4)柱温的考察
[0125]
取同一批次锁阳待测样品(批号190801-748100-02),按照实施例1中的方法制成3份供试品溶液,分别在不同柱温(33℃、35℃和37℃)下按照实施例1中的色谱条件,测定。以4号峰为参照峰,计算色谱图中共有峰的相对保留时间和相对峰面积,见表6和表7。结果显示,7号峰和8号峰的保留时间和峰面积受柱温影响波动较大,其余共有峰相对保留时间rsd值均小于1.9%,表明该方法的柱温耐用性较好。
[0126]
表6 特征峰的保留时间和相对保留时间
[0127][0128]
表7 特征峰的峰面积和相对峰面积
[0129][0130][0131]
(5)流速的考察
[0132]
取同一批次锁阳待测样品(批号190801-748100-02),按照实施例1中的方法制成3份供试品溶液,分别在不同流速下(0.29ml/min、0.30ml/min和0.31ml/min)按照实施例1中的色谱条件,测定。以4号峰为参照峰,计算色谱图中共有峰的相对保留时间和相对峰面积,见表8和表9。结果显示,5号峰的峰面积对流速变化较敏感,共有峰相对保留时间rsd值均不大于2.2%,表明该方法的流速耐用性较好。
[0133]
表8 特征峰的保留时间和相对保留时间
84%;21-25min,流动相a:流动相b为16-18%:84-82%;25-26min,流动相a:流动相b为18%:82%。
[0142]
实验组1、对照组1-3得到的特征图谱见图7-10,从图中可以看出,对照组1得到的特征图谱的特征峰的分离效果和峰形较差;对照组2得到的特征图谱的特征峰分离效果较差,1号峰和2号峰分离不明显,对照组3得到的特征图谱的基线不平稳,部分非特征峰分离效果差。由此可见,本发明采用的梯度洗脱程序得到的特征图谱的特征峰的分离度佳、峰形较好。
[0143]
实验例3流动相考察
[0144]
取同一批次的锁阳待测样品(批号190801-015000-01),按照实施例1中的方法制成8份供试品溶液,然后按照实施例1中的色谱条件,测定,区别仅在于:
[0145]
实验组1,以甲醇为流动相a,以水为流动相b;
[0146]
实验组2,以乙腈为流动相a,以水流动相b;
[0147]
实验组3,以甲醇为流动相a,以体积分数0.05%磷酸溶液流动相b;
[0148]
实验组4,以甲醇为流动相a,以体积分数0.05%甲酸溶液流动相b;
[0149]
实验组5,以甲醇为流动相a,以体积分数0.1%磷酸溶液流动相b;
[0150]
实验组6,以甲醇为流动相a,以体积分数0.1%甲酸溶液流动相b;
[0151]
实验组7,以甲醇为流动相a,体积分数0.2%甲酸溶液为流动相b;
[0152]
实验组8,以甲醇为流动相a,以体积分数0.2%乙酸溶液流动相b;
[0153]
当流动相a为甲醇,流动相b为体积分数0.2%甲酸溶液时,色谱峰的峰形较好,分离度较高,色谱峰分布均匀。
[0154]
实验例4待测样品的考察
[0155]
实验组1:取锁阳药材待测样品(批号yc190801-748100-02),研细,取1.0g,精密称定,置于具塞锥形瓶中,精密加入体积分数30%甲醇10ml,密塞,称定重量,超声(功率500w,频率40kh)处理20min,放至室温,补足减失的重量,摇匀,离心5min,转速为3500转,然后取上清液,过0.22μm微孔滤膜,得到浓度为1g/ml的供试品溶液。
[0156]
实验组2:取锁阳饮片待测样品(批号yp190801-748100-02),研细,取1.0g,精密称定,置于具塞锥形瓶中,精密加入体积分数30%甲醇10ml,密塞,称定重量,超声(功率500w,频率40kh)处理20min,放至室温,补足减失的重量,摇匀,离心5min,转速为3500转,然后取上清液,过0.22μm微孔滤膜,得到浓度为1g/ml的供试品溶液。
[0157]
实验组3:取锁阳配方颗粒待测样品(批号kl190801-748100-02),研细,取1.0g,精密称定,置于具塞锥形瓶中,精密加入体积分数30%甲醇10ml,密塞,称定重量,超声(功率500w,频率40kh)处理20min,放至室温,补足减失的重量,摇匀,离心5min,转速为3500转,然后取上清液,过0.22μm微孔滤膜,得到浓度为1g/ml的供试品溶液。
[0158]
实验组1-3制得的供试品溶液按照实施例1“色谱条件”进样,注入量为2μl,测定,得到特征图谱,见图11(s1-s3依次代表锁阳药材、锁阳饮片、锁阳颗粒),相对保留时间见表10。
[0159]
表10 锁阳药材、饮片、颗粒特征峰的相对保留时间
[0160]
名称峰1峰2峰3峰4(s)峰5峰6峰7峰8锁阳药材0.5720.6440.8771.0002.4292.8143.1253.296
锁阳饮片0.4720.5410.7931.0002.4102.8413.3713.545锁阳配方颗粒0.4670.5340.8081.0002.3542.7443.3103.482标准图谱0.470.540.811.002.392.823.353.52
[0161]
表10和图11中可以看出,本发明提供的锁阳及其制剂特征图谱的构建方法适用于锁阳药材、锁阳饮片、锁阳配方颗粒和锁阳标准汤剂冻干粉。
[0162]
实施例2
[0163]
本实施例提供了一种锁阳中原儿茶酸含量的检测方法,包括以下步骤,
[0164]
对照品溶液的制备:取原儿茶酸对照品,加入体积分数50%甲醇水溶液,制备系列浓度不同的原儿茶酸对照品溶液,浓度分别是0.1998mg/ml、0.0799mg/ml、0.0200mg/ml、0.0080mg/ml和0.0040mg/ml。
[0165]
供试品溶液的制备:取锁阳待测样品1.0g,精密称定,置于具塞锥形瓶中,精密加入体积分数30%甲醇10ml,密塞,称定重量,超声(功率500w,频率40kh)处理20min,放至室温,补足减失的重量,摇匀,离心5min,转速为3500转,然后取上清液,过0.22μm微孔滤膜,得到浓度为1g/ml的供试品溶液。
[0166]
含量测定:采用超高效液相色谱仪测定对照品溶液和供试品溶液中原儿茶酸的峰面积,根据外标法计算供试品溶液中原儿茶酸的含量。
[0167]
其中,色谱条件为:按照高效液相色谱法测定,以waters其中,色谱条件为:按照高效液相色谱法测定,以waterst3(2.1
×
100mm,1.6μm),以甲醇为流动相a,以体积分数0.2%甲酸溶液为流动相b,按照梯度洗脱程序进行色谱分析,梯度洗脱程序为:0-9min,流动相a:流动相b为0%:100%;9-10min,流动相a:流动相b为0-10%:100-90%;10-13min,流动相a:流动相b为10%:90%;13-20min,流动相a:流动相b为10-14%:90-86%;20-26min,流动相a:流动相b为14-16%:86-84%;26-30min,流动相a:流动相b为16-18%:84-82%,流动相流速为0.3ml/min,检测波长为260nm;柱温35℃。
[0168]
标准曲线的制定:以原儿茶酸色谱峰的峰面积为纵坐标,以原儿茶酸对照品溶液的浓度为横坐标,原儿茶酸对照品溶液的浓度和峰面积见表11,以最小二乘法进行线性回归,得到回归方程:y=23325x-12171.6,r=0.9999,线形范围为0.0040-0.1998mg/ml,原儿茶酸的浓度与峰面积的标准曲线见图12。图13是原儿茶酸对照品溶液的特征图谱。
[0169]
表11 原儿茶酸的浓度和对应的峰面积
[0170]
原儿茶酸浓度(mg/ml)0.19980.07990.02000.00800.0040原儿茶酸峰面积4643085186849143868717293486158
[0171]
取10个批次锁阳标准汤剂冻干粉,按照上述供试品溶液的制备制成10个批次供试品溶液,按照上述方法测定供试品溶液中原儿茶酸的峰面积,供试品溶液的色谱图见图14;通过外标法定量,得到供试品溶液中原儿茶酸的含量,每个样品测试两次,结果见表12。
[0172]
表12 各个供试品溶液中原儿茶酸的含量
[0173][0174][0175]
实验例5锁阳中原儿茶酸含量测定方法学考察
[0176]
(1)溶剂的影响
[0177]
取2μl体积分数30%甲醇溶剂注入超高效液相色谱仪,按照实施例2中的色谱条件,测定,得到的色谱图中无色谱峰,说明溶剂对锁阳特征图谱的色谱峰没有影响,色谱图见图15。
[0178]
(2)重复性实验
[0179]
取同一批次锁阳待测样品(批号190801-748100-02),按照实施例2中的方法制成6份供试品溶液,按照实施例2中的方法测定供试品溶液中的原儿茶酸含量。结果见表13,原儿茶酸测定结果的rsd值为2.1%,符合《中国药典》2015版四部9101(药品质量标准分析方法验证指导原则)关于方法重复性的要求。
[0180]
表13 原儿茶酸重复性实验结果
[0181]
序号取样量(g)峰面积含量(mg/g)11.00024351290.23021.00074250450.22531.00024383310.23241.00044411320.23450.99994442810.23560.99984526870.240rsd(%)//2.1
[0182]
(3)精密度实验
[0183]
取同一批次锁阳待测样品(批号190801-748100-02),按照实施例2中的方法制成6份供试品溶液,由不同的分析人员,利用另外一台waters超高效液相色谱仪按照实施例2中的方法测定供试品溶液中原儿茶酸的含量,结果见表14。结果显示,中间精密度实验所测得的原儿茶酸含量的rsd值为0.5%,与重复性检测结果的rsd值为1.15%,符合《中国药典》2015版四部9101(药品质量标准分析方法验证指导原则),说明本发明提供的锁阳中原儿茶酸含量的测定方法的精密度良好。
[0184]
表14 中间精密度实验结果
[0185][0186][0187]
(4)准确度实验
[0188]
取原儿茶酸对照品,精密称定,加入体积分数50%甲醇制成浓度为0.1mg/ml的对照品溶液。取“重复性实验”项下已知含量的供试品9份(190801-748100-02),分成3组,每组3份,每份供试品溶液1.0g,精密称定,具体数值见表15,3组供试品溶液中分别精密加入含有原儿茶酸0.0999mg、0.1998mg、0.2997mg的对照品溶液10ml,按照实施例2中锁阳中原儿茶酸含量的检测方法进行检测,按照式ⅰ计算回收率,结果见表15。结果说明,回收率范围为97.3-108.1%,平均回收率为104.3%,rsd值为4.0%,符合《中国药典》2015年版四部9101(药品质量标准分析方法验证指导原则)的规定,表明所建立含量测定方法的准确性良好。
[0189][0190]
表15 回收率实验结果
[0191]
[0192]
(5)稳定性实验
[0193]
取同一批次的锁阳待测样品(批号190801-748100-02),按照实施例2中的方法制成供试品溶液,分别于室温条件下放置0h、2h、4h、6h、8h、10h、12h、24h后,按照实施例2中的方法测定供试品溶液中原儿茶酸的含量,结果见表16。结果显示,原儿茶酸含量在12h内的rsd值<2.0%,表明供试品溶液在12h内稳定。
[0194]
表16 稳定性实验结果
[0195][0196]
(6)柱温的考察
[0197]
取同一批次锁阳待测样品(批号190801-748100-02),按照实施例2中的方法制成6份供试品溶液,分别于不同柱温条件下按照实施例2中的方法测定供试品溶液中的原儿茶酸含量,柱温分别是33℃、35℃和37℃,每个温度重复两次实验,结果见表17,原儿茶酸测定结果的rsd值为2.4%,基本符合《中国药典》2015版四部9101(药品质量标准分析方法验证指导原则)关于方法耐用性的要求。
[0198]
表17 不同柱温考察实验结果
[0199][0200]
(7)流速的考察
[0201]
取同一批次锁阳待测样品(批号190801-748100-02),按照实施例2中的方法制成6份供试品溶液,分别于不同流速条件下按照实施例2中的方法测定供试品溶液中的原儿茶酸含量,流速分别是0.29ml/min、0.30ml/min和0.31ml/min,每个流速重复两次实验,结果见表18,原儿茶酸测定结果的rsd值为2.8%,基本符合《中国药典》2015版四部9101(药品质量标准分析方法验证指导原则)关于方法耐用性的要求。
[0202]
表18 不同流速考察实验结果
[0203][0204][0205]
显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或
变动。这里无需也无法对所有的实施方式予以穷举。而由此所引申出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1