逍遥丸的指纹图谱检测方法与流程
1.本发明属于中成药检测技术领域,具体地说,涉及一种逍遥丸的指纹图谱检测方法。
背景技术:
2.逍遥丸源于宋代汉医方剂学著作—《太平惠民和剂局方》,是由柴胡、当归、白芍、炒白术、茯苓、炙甘草和薄荷等共七味药材制成的成方制剂,临床用于治疗肝郁脾虚所致的郁闷不舒、胸胁胀痛、头晕目眩、食欲减退和月经不调等疾病。《中国药典》2015版中逍遥丸以芍药苷含量为指标控制其质量
1.,国家部颁标准中则是以当归和白芍的鉴别反应
2.为质量控制标准。
3.逍遥丸在中医临床中应用广泛,因此相关学者也通过检测逍遥丸部分有效成分的途径来完成质量控制与评价,其中,徐宇等以柴胡和甘草的鉴别反应结合芍药苷含量作为其控制标准,高晶等应用hplc法测定槲皮素含量,刘幸平等测定阿魏酸的含量,韩慧琴等测定甘草含量,苏兰宜等检测芍药苷和槲皮素含量,田洁等同时测定阿魏酸和甘草含量,梁晓东等测定芍药苷,为逍遥丸的质量控制提供思路。
4.但是,目前对逍遥丸中主要成分的含量测定主要集中在芍药苷这一主要有效成分,检测指标单一,故本实验拟对芍药苷、甘草苷,白术内酯、藁本内酯、甘草酸等多种成分完成一测多评体系下的含量测定。建立逍遥丸hplc指纹图谱方法,指认主要成分,可以为全面评价和控制逍遥丸质量及临床应用提供参考依据。
技术实现要素:
5.发明目的:本发明的目的是为了克服现有技术的不足,开发一种关于逍遥丸的指纹图谱检测方法,通过本发明的方法可以客观,正确,有效的控制逍遥丸的质量,对确保其安全性与有效性,保证临床质量具有重要的指导意义。
6.技术方案:为实现以上目的,本发明采用的技术方案为:
7.一种逍遥丸的指纹图谱检测方法,它包括以下步骤:
8.步骤1、逍遥丸供试品溶液的制备:
9.取逍遥丸样品适量,研细,粉末过筛,置于具塞锥形瓶中,精密加入甲醇,浸渍后超声处理,放冷,摇匀,滤过,取续滤液,经0.45μm微孔滤膜滤过,置于容量瓶,甲醇定容至刻度线,即得逍遥丸供试品溶液;
10.步骤2、逍遥丸对照品溶液的制备:
11.分别精密称取甘草苷、芍药苷、绿原酸、异甘草素、甘草酸铵、甘草次酸、芍药内酯苷、白术内酯、藁本内酯对照品,加甲醇分别制成对照品原液,精密吸取上述各成分对照品,置于容量瓶中,即得混合对照品溶液;
12.步骤3、对照品线性关系的建立
13.步骤4、分别精密吸取按步骤1方法得到的逍遥丸供试品溶液和步骤2的混合对照
品溶液,注入高效液相色谱仪,记录色谱图;
14.步骤5、将步骤3中获得的逍遥丸供试品溶液的指纹图谱导出,并导入中药色谱指纹图谱相似度评价系统2004a;选择不同批次逍遥丸的色谱图中均存在的色谱峰作为共有峰;用平均值计算法生成逍遥丸的对照指纹图谱,计算各共有峰的相对保留时间和相对峰面积;并根据混合对照品溶液色谱图的保留时间标注对照指纹图谱中峰的化学成分。
15.作为优选方案,以上所述的一种逍遥丸的指纹图谱检测方法,
16.步骤1、逍遥丸供试品溶液的制备方法为:
17.取逍遥丸样品适量,研细,粉末过50目筛,约1g,置于具塞锥形瓶中,精密加入甲醇10ml,浸渍30min后超声处理30min,放冷,摇匀,滤过,取续滤液,经0.45μm微孔滤膜滤过,置于10ml容量瓶,甲醇定容至刻度线,即得逍遥丸供试品溶液。
18.作为优选方案,以上所述的一种逍遥丸的指纹图谱检测方法,其特征在于,
19.步骤2、混合对照品溶液的制备方法为:
20.分别精密称取甘草苷、芍药苷、绿原酸、异甘草素、甘草酸铵、甘草次酸、芍药内酯苷、白术内酯和藁本内酯对照品,加甲醇分别制成浓度为3mg/ml的对照品原液;精密吸取上述各对照品原液各100μl,置于10ml容量瓶中,加甲醇定容,即得混合对照品溶液。
21.作为优选方案,以上所述的一种逍遥丸的指纹图谱检测方法,其特征在于,
22.步骤4、高效液相色谱仪检测的色谱条件为:
23.色谱柱:ymc-pack ods-a,规格250mm
×
4.6mm,5μm;流动相为乙腈-0.1%磷酸水溶液;梯度洗脱:0.01~5min,5%乙腈;5~15min,5%~15%乙腈;15~45min,15%~35%乙腈;45~63min,35%~70%乙腈;63~75min,70%~76%乙腈;75~87min,76%~100%乙腈;体积流量为0.8ml/min;柱温30℃;进样量为10μl;检测波长为254nm。
24.作为优选方案,以上所述的一种逍遥丸的指纹图谱检测方法,
25.步骤3线性关系建立为:
26.精密吸取步骤(2)的混合对照品溶液2ml,1.5ml,1ml,0.5ml,0.2ml,0.1ml分别置于2ml量瓶中,用甲醇定容,即得不同梯度的混合对照品溶液,依次注入高效液相色谱仪;分别以对照品的峰面积y和浓度x建立九个成分的线性回归方程;其中,绿原酸线性回归方程为y=1e+07x+151750,r=0.9999;芍药内酯苷线性回归方程为y=928150x+243140,r=0.9997;芍药苷线性回归方程为y=2e+06x+25598,r=0.9999;甘草苷线性回归方程为y=5e+06x+488841,r=0.9990;甘草酸铵线性回归方程为y=5e+06x-781599,r=0.9999;异甘草素线性回归方程为y=2e+07x+15081,r=0.9999;藁本内酯线性回归方程为y=1e+07x+6919.7,r=0.9995;白术内酯线性回归方程为y=4e+07x+23050,r=0.9999;甘草次酸线性回归方程为y=2e+07x+18419,r=0.9999。
27.作为优选方案,以上所述的一种逍遥丸的指纹图谱检测方法,本发明共建立17个共有峰并指认了9种成分,其中5号峰为绿原酸、6号峰为芍药内酯苷、7号峰为芍药苷、8号峰为甘草苷、11号峰为甘草酸铵、12号峰为异甘草素、15号峰为藁本内酯、16号峰为白术内酯、17号峰为甘草次酸。
28.提取与分析方法筛选考察
29.本实验考察了超声30min、45min、60min以及回流提取2h的提取方法,其中,超声提取30min,提取效果较为完全且具有效率高能耗低简便易操作的优点,故选择超声提取
30min作为提取方法。
30.通过实验比较可知,在乙腈-水、甲醇-水、乙腈-0.1%磷酸水三种流动相系统中,逍遥丸提取物在乙腈-0.1%磷酸水这一流动相系统中,出峰较多且各峰峰形分离度较好,故选择乙腈-0.1%磷酸水流动相系统。
31.确定最佳流动相之后,本发明又筛选了不同的梯度洗脱程序,根据出峰情况及各峰分离度效果,确定最佳洗脱梯度梯度洗脱为:0.01~5min,5%乙腈;5~15min,5%~15%乙腈;15~45min,15%~35%乙腈;45~63min,35%~70%乙腈;63~75min,70%~76%乙腈;75~87min,76%~100%乙腈。
32.本发明的有益效果是:
33.(1)本发明通过大量实验筛选出最佳的供试品制备方法与对照品的制备方法和最佳的色谱分析条件,建立的逍遥丸指纹图谱和各对照品化合物的标准曲线,不但能有效地表征逍遥丸的质量,且能准确的检测各对照品化合物的含量,有利于全面监控逍遥丸药物的质量。
34.(2)本发明方法具有稳定性好、精密度高、准确度高、重现性好等的优点。可以全面、客观准确的评价和控制逍遥丸的质量。
附图说明
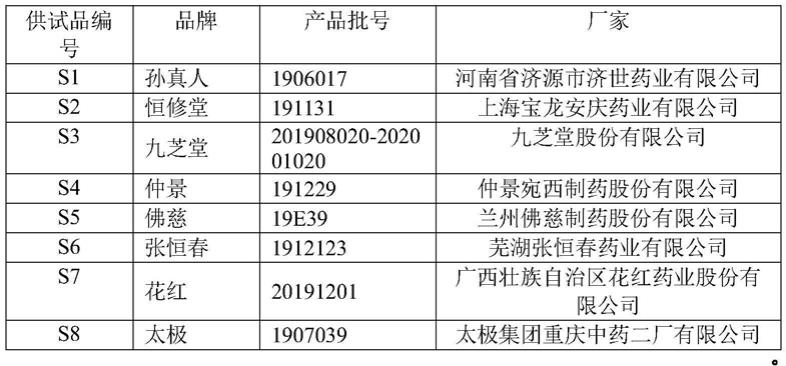
35.图1为混合对照品溶液的hplc的的色谱图。
36.图2为供试品溶液的hplc的的色谱图。
37.图3为8批逍遥丸的指纹图谱。
具体实施方式
38.下面将结合实施例对本发明的实施方案进行详细描述,实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。
39.实施例1
40.以下实施例所用仪器
41.实验仪器
42.岛津ic-20at型高效液相色谱仪(spd-m20a二极管阵列检测器),bp211d型万分之一精确分析天平(赛多利斯),fa1004n型十万分之一精确电子分析天平(赛多利斯),hh-6型数字显视恒温水浴锅(常州市江南实验仪器厂),shz-d(iii)型循环水式真空泵(巩义市予华仪器有限责任公司),移液枪(100-1000μl,大龙兴创仪器),kq5200de型数控超声波清洗器(频率:40khz,昆山市超声仪器有限公司)。camag薄层色谱扫描仪(linomat5)、camag tlc visualizer薄层色谱成像及文件系统,中药色谱指纹图谱相似度评价系统a版。
43.1.2实验试剂及材料
44.茯苓对照药材(批号121117-200403,购自中国药品生物制品检定所)、白术对照药材(批号120925-200708,购自中国药品生物制品检定所)、异甘草素(hplc≥98%,批号190311,购自南京森贝伽生物科技有限公司)、当归对照药材(批号120927-200613,购自中国药品生物制品检定所)、白芍对照药材(批号120905-200508,购自中国药品生物制品检定
所)、薄荷对照药材(批号120916-201109,购自中国食品药品检定研究院)、芍药苷(hplc≥98%,批号pcl-#-p857,购自南京森贝伽生物科技有限公司)、甘草苷(hplc≥98%,批号:180808,购自南京森贝伽生物科技有限公司)、绿原酸(hplc≥98%,批号:160527,购自南京森贝伽生物科技有限公司)、柴胡对照药材(批号c-011-130517,购自成都瑞芬思生物科技有限公司)、甘草对照药材(批号120904-201620,购自中国食品药品检定研究所)、白术内酯(hplc≥98%,批号111975-201501,购自南京森贝伽生物科技有限公司)、芍药内酯苷(hplc≥98%,批号s-011-130506,购自成都瑞芬思生物科技有限公司))、甘草酸铵(hplc≥98%,购自上海同田生物技术有限公司)、甘草次酸(hplc≥98%,批号723-8601,购自卫生部药品生物制品检定所)、藁本内酯(hplc≥98%,批号111737-201507,购自中国食品药品检定研究院),水为纯化水,甲醇、乙腈为色谱纯,其余试剂均为分析纯。逍遥丸样品来源见表1。
45.表1逍遥丸(浓缩丸)编号、品牌、产品批号、产商
[0046][0047]
实施例1
[0048]
一种逍遥丸的指纹图谱检测方法,它包括以下步骤:
[0049]
步骤1、逍遥丸供试品溶液的制备:
[0050]
取以上8批次的逍遥丸样品适量,研细,粉末过50目筛,各1g,分别置于具塞锥形瓶中,分别精密加入甲醇10ml,浸渍30min后超声处理30min,放冷,摇匀,滤过,取续滤液,经0.45μm微孔滤膜滤过,置于10ml容量瓶,甲醇定容至刻度线,即得8批次的逍遥丸供试品溶液;
[0051]
步骤2、逍遥丸对照品溶液的制备:
[0052]
分别精密称取甘草苷、芍药苷、绿原酸、异甘草素、甘草酸铵、甘草次酸、芍药内酯苷、白术内酯和藁本内酯对照品,加甲醇分别制成浓度为3mg/ml的对照品原液;精密吸取上述各对照品原液各100μl,置于10ml容量瓶中,加甲醇定容,即得混合对照品溶液;
[0053]
步骤3、对照品线性关系的建立
[0054]
精密吸取步骤(2)的混合对照品溶液2ml,1.5ml,1ml,0.5ml,0.2ml,0.1ml分别置于2ml量瓶中,用甲醇定容,即得不同梯度的混合对照品溶液,依次注入高效液相色谱仪;分别以对照品的峰面积y和浓度x建立九个成分的线性回归方程;其中,绿原酸线性回归方程为y=1e+07x+151750,r=0.9999;芍药内酯苷线性回归方程为y=928150x+243140,r=0.9997;芍药苷线性回归方程为y=2e+06x+25598,r=0.9999;甘草苷线性回归方程为y=5e+06x+488841,r=0.9990;甘草酸铵线性回归方程为y=5e+06x-781599,r=0.9999;异甘
草素线性回归方程为y=2e+07x+15081,r=0.9999;藁本内酯线性回归方程为y=1e+07x+6919.7,r=0.9995;白术内酯线性回归方程为y=4e+07x+23050,r=0.9999;甘草次酸线性回归方程为y=2e+07x+18419,r=0.9999。
[0055]
步骤4、分别精密吸取按步骤1方法得到的逍遥丸供试品溶液和步骤2的混合对照品溶液,注入高效液相色谱仪,记录色谱图;混合对照品溶液的hplc的的色谱图如图1所示。供试品溶液的hplc的的色谱图如图2所示。8批逍遥丸的指纹图谱如图3所示。
[0056]
供试品高效液相色谱仪检测的色谱条件为:
[0057]
色谱柱:ymc-pack ods-a,规格250mm
×
4.6mm,5μm;流动相为乙腈-0.1%磷酸水溶液;梯度洗脱:0.01~5min,5%乙腈;5~15min,5%~15%乙腈;15~45min,15%~35%乙腈;45~63min,35%~70%乙腈;63~75min,70%~76%乙腈;75~87min,76%~100%乙腈;体积流量为0.8ml/min;柱温30℃;进样量为10μl;检测波长为254nm。
[0058]
步骤5、将步骤3中获得的逍遥丸供试品溶液的指纹图谱导出,并导入中药色谱指纹图谱相似度评价系统2004a;选择不同批次逍遥丸的色谱图中均存在的色谱峰作为共有峰;用平均值计算法生成逍遥丸的对照指纹图谱,计算各共有峰的相对保留时间和相对峰面积;并根据混合对照品溶液色谱图的保留时间标注对照指纹图谱中峰的化学成分。得到17个共有峰并指认了9种成分,其中5号峰为绿原酸、6号峰为芍药内酯苷、7号峰为芍药苷、8号峰为甘草苷、11号峰为甘草酸铵、12号峰为异甘草素、15号峰为藁本内酯、16号峰为白术内酯、17号峰为甘草次酸。
[0059]
用生成的对照指纹图谱r来建立共有模式,测得除s1孙真人、s2恒修堂外,6批逍遥丸和其共有模式之间相似度均在0.9以上,说明建立的指纹图谱可以作为逍遥丸质量评价的标准之一,相似度结果见表2。
[0060]
表2 8批次逍遥丸相似度结果
[0061][0062]
含量测定
[0063]
取6批次表1的九芝堂逍遥丸适量,按上述方法制备得到供试品溶液,按以上色谱条件进样测定,依据上述步骤3的线性回归方程,计算甘草苷、芍药苷、绿原酸、异甘草素、甘草酸铵、甘草次酸、白术内酯及藁本内酯的含量,结果见表3。6个批次9种成分含量的rsd值较小,说明不同批次逍遥丸之间质量较为稳定。
[0064]
表3 9种成分的含量测定结果(mg/g)
[0065][0066]
实施例2指纹图谱检测的方法学研究:
[0067]
1、方法学考察
[0068]
1.1精密度实验
[0069]
照实施例1制备一份供试品溶液,按照实施例1所述的色谱条件,连续进样6次,记录9组对照峰的保留时间和峰面积,计算可得各组成分峰面积和保留时间的rsd均小于3%,结果表明本方法精密度良好。
[0070]
1.2重复性实验
[0071]
照实施例1制备一份供试品溶液,按照实施例1所述的色谱条件进样检测,记录9组对照峰的峰面积以及保留时间,计算可得各组成分的峰面积和保留时间的rsd均小于3%,结果表明本方法重复性良好。
[0072]
1.3稳定性实验
[0073]
照实施例1制备平行制备6份供试品溶液,分别于0h,2h,4h,8h,12h,24h按照实施例1所述的色谱条件进样检测,记录9个成分的峰面积及保留时间,并计算rsd。计算可得各组成分峰面积和保留时间的rsd均小于3%,结果表明供试品在24小时内稳定性良好。
[0074]
1.4加样回收实验
[0075]
精密称取已知含量样品6份,精密称定,置于具塞锥形瓶中。分别精密称取甘草苷1.15mg、芍药苷0.61mg、绿原酸1.60mg、异甘草素0.04mg、甘草酸铵1.11mg、甘草次酸0.03mg、芍药内酯苷0.27mg、白术内酯0.02mg、藁本内酯0.06mg。照实施例1制备供试品溶液,按照实施例1所述的下色谱条件进样检测。计算9个成分的加样回收率,结果见表4。
[0076]
表4 9个成分的加样回收率
[0077]
[0078]
[0079][0080]
1.5专属性实验
[0081]
取逍遥丸样品溶液、甘草苷、芍药苷、绿原酸、异甘草素、甘草酸铵、甘草次酸、芍药内酯苷、白术内酯、藁本内酯对照品溶液以及缺柴胡、当归、白芍、炒白术、茯苓、炙甘草和薄荷的阴性样品溶液,按照实施例1的下方法进样分析,样品溶液中甘草苷、芍药苷、绿原酸、异甘草素、甘草酸铵、甘草次酸、芍药内酯苷、白术内酯、藁本内酯均分离度较好,且阴性样品中在各待测成分相应保留时间处无吸收峰干扰,说明该方法专属性符合要求。
[0082]
由以上实验结果表明,本发明建立的逍遥丸指纹图谱检测方法,精密度、稳定性和重复性好,能有效地表征逍遥丸的质量,有利于全面监控其质量。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1