一种检测水产品中呈味肽L-γ-谷氨酰-L-缬氨酰-甘氨酸的方法与流程
本发明涉及食品检测技术领域,特别涉及一种检测水产品中呈味肽l-γ-谷氨酰-l-缬氨酰-甘氨酸的方法。
背景技术:
γ-谷氨酰肽是指含有γ-谷氨酰残基的一类小分子肽,是由谷氨酸或者谷氨酰胺的γ-羧基基团与氨基酸或多肽的氨基基团发生脱水缩合形成的,近年来,因其具有显著的呈味效果而被广泛关注。γ-谷氨酰肽作为kokumi风味(又称浓厚味)的主要贡献者,赋予食物特有的浓厚感、持续性、饱满感和丰富度,并且会提升五种基本口味的协调性以及咸味、鲜味和甜味的呈味强度。l-γ-谷氨酰-l-缬氨酰-甘氨酸(γ-glu-val-gly)是γ-谷氨酰肽中呈味强度最高的物质,研究表明,其味感受活性是谷胱甘肽的12.8倍。kuroda团队陆续在商业鱼露、酱油、虾酱、酿造酒精饮料、扇贝及加工扇贝等食品中检测鉴定获得γ-glu-val-gly,sofyanovich等也在酵母提取物中分离获得该三肽物质。
目前关于γ-glu-val-gly的测定方法主要有高效液相色谱法和液相色谱串联质谱法。食品安全标准与监测评估司发布的《关于弯曲乳杆菌等24种“三新食品”的公告(2019年第2号)》中确定了适用于通过化学合成制得的食品添加剂γ-glu-val-gly的测定方法。该方法以高效液相色谱进行检测,采用ods-c18(4.6×250mm,5μm)色谱柱和紫外检测器,根据国家标准gb/t27579-2011《精油高效液相色谱分析通用法》中9.0内标法进行定量。但该方法灵敏度不高,不能完全满足痕量或超痕量检测。已有文献关于γ-glu-val-gly的检测多采用lc-ms/ms法,即以6-氨基喹啉基-n-羟基琥珀酰亚氨基甲酸酯(aqc)作为柱前衍生试剂,通过反相高效液相色谱对样品进行分离,利用ms/ms进行结构鉴定,质谱检测采用电喷雾离子源正离子扫描模式,多反应监测(mrm)模式下对碎片化的目标物进行特定质荷比扫描,以内标法进行定量分析。但该方法的缺陷在于其专用aqc衍生试剂盒价格昂贵,并且aqc衍生试剂易与环境中的潮气反应,稳定性差,保存和使用过程中易被污染失效;另外,据kuroda团队文献报道,以aqc为衍生化试剂获得的样品中γ-glu-val-gly衍生物色谱图复杂(图7),衍生化副产物对目标物的定性定量分析产生干扰。
技术实现要素:
有鉴于此,本发明目的在于提供一种检测水产品中呈味肽l-γ-谷氨酰-l-缬氨酰-甘氨酸的方法,本发明以稳定性好且价格低廉的2,4-二硝基氯苯为衍生剂进行呈味肽l-γ-谷氨酰-l-缬氨酰-甘氨酸的检测,具有准确度高、灵敏度高、成本低的优点;且多反应监测模式下获得目标物定性、定量离子色谱图简单,无衍生副产物的干扰。
为了实现上述发明目的,本发明提供以下技术方案:
本发明提供了一种检测水产品中呈味肽l-γ-谷氨酰-l-缬氨酰-甘氨酸的方法,包括以下步骤:
(1)提供待测水产品提取液,将所述待测水产品提取液与内标物溶液和去离子水混合,得到待测混合液;
(2)将所述待测混合液与缓冲溶液、2,4-二硝基氯苯溶液混合进行涡旋,得到待测涡旋液;将所述待测涡旋液在50~90℃条件下避光进行衍生化反应,得到待测进样液;
(3)将所述待测进样液进行液相色谱-串联质谱检测,得到γ-glu-val-gly色谱图和内标物色谱图;
(4)将由所述γ-glu-val-gly色谱图和内标物色谱图得到的γ-glu-val-gly和内标物的峰面积之比代入标准曲线,计算得到水产品中γ-glu-val-gly的含量;所述标准曲线为γ-glu-val-gly标准品和内标物的峰面积之比与γ-glu-val-gly标准品溶液浓度之间的线性曲线。
优选地,所述步骤(1)中提供待测水产品提取液的方法包括:
将待测水产品的可食部位剪碎后进行研磨,得到匀浆样品;
将所述匀浆样品与去离子水混合,将所得混合料液依次进行均质、搅拌和离心,得到上清液;
将所述上清液经0.45μm滤膜过滤,再将滤液进行超滤离心,所得下层滤液为所述待测水产品提取液。
优选地,所述匀浆样品与去离子水的用量比为10g:100ml;所述均质和搅拌均在冰浴条件下进行;所述均质的速度为9000rpm,时间为3min;所述搅拌的速度为300rpm,时间为20min;
所述离心和超滤离心的温度为4℃,离心力为5000g,离心和超滤离心的时间独立为10~15min;所述超滤离心的滤膜截留分子量为10kd。
优选地,所述步骤(1)中的内标物为l-精氨酸-15n4;所述内标物溶液的浓度为0.2mg/l;所述待测水产品提取液、内标物溶液与去离子水的体积比为1:1~2:1。
优选地,所述步骤(2)中的缓冲溶液为na2co3-nahco3溶液,所述缓冲溶液的ph值为9.0;所述缓冲溶液与待测混合液的体积比为1:1~2:1。
优选地,所述步骤(2)中2,4-二硝基氯苯溶液为2,4-二硝基氯苯的乙腈溶液;所述2,4-二硝基氯苯溶液的质量浓度为5~20%;所述2,4-二硝基氯苯溶液与待测混合液的体积比为1:1~1:3。
优选地,所述步骤(2)中衍生化反应的时间为60~100min;所述衍生化反应后加入乙酸终止反应。
优选地,所述衍生化反应终止后,还包括将所得反应液与水混合定容,然后经0.45μm滤膜过滤,滤液为所述待测进样液。
优选地,所述步骤(3)液相色谱-串联质谱检测中液相色谱条件包括:
c18色谱柱:粒径3μm,尺寸规格2.1mmid×100mm;柱温:35℃;
流动相:a相为25mm甲酸水溶液,ph值为6.0;b相为60%体积分数的乙腈水溶液;流动相梯度洗脱程序:0~6min,b相体积分数35%;6.1~12min,b相体积分数100%;12.1~20min,b相体积分数35%;
进样量:2.0μl;流速:0.2ml/min。
优选地,所述步骤(3)液相色谱-串联质谱检测中质谱条件包括:
电喷雾离子源为正离子扫描模式;
离子源参数:雾化气温度350℃,雾化气流速为12l/min,雾化器压力为20psi,毛细管电压为4.0kv;
一级质谱数据采集采用全扫描模式,扫描范围为m/z50~500;二级质谱数据采集采用多反应监测模式;
γ-glu-val-gly的定性离子对为470.1/395、470.1/367和470.1/184.2、定量离子对为470.1/72.2,内标物定量离子对为345.0/71.1。
本发明提供了一种检测水产品中呈味肽l-γ-谷氨酰-l-缬氨酰-甘氨酸的方法,包括以下步骤:(1)提供待测水产品提取液,将所述提取液与内标物溶液和去离子水混合,得到待测混合液;(2)将所述待测混合液与缓冲溶液、2,4-二硝基氯苯溶液混合进行涡旋,得到待测涡旋液;将所述待测涡旋液在50~90℃条件下避光进行衍生化反应,得到待测进样液;(3)将所述待测进样液进行液相色谱-串联质谱检测,得到γ-glu-val-gly谱图和内标物谱图;(4)将由所述γ-glu-val-gly谱图和内标物谱图得到的γ-glu-val-gly和内标物的峰面积之比代入标准曲线,计算得到水产品中γ-glu-val-gly的含量;所述标准曲线为γ-glu-val-gly标准品和内标物的峰面积之比与γ-glu-val-gly标准品溶液浓度之间的线性曲线。本发明采用2,4-二硝基氯苯为衍生试剂,γ-glu-val-gly与2,4-二硝基氯苯(cdnb)之间发生亲核芳香取代反应,亲核阴离子由γ-glu-val-gly碱基电离生成,它攻击2,4-二硝基氯苯中含氯原子的碳,使氯离子离开从而生成γ-glu-val-gly衍生产物。本发明采用的2,4-二硝基氯苯衍生试剂不仅稳定性好,而且价格低廉,不需要专门的衍生试剂盒,以所述衍生试剂进行呈味肽l-γ-谷氨酰-l-缬氨酰-甘氨酸的检测,具有准确度高、灵敏度高、成本低的优点;而且,多反应监测模式下获得目标物定性、定量离子色谱图简单,无衍生副产物的干扰,可以满足水产品中呈味多肽l-γ-谷氨酰-l-缬氨酰-甘氨酸的定性、定量检测需求;并且操作简便。
实施例结果表明,本发明提供的检测水产品中呈味肽l-γ-谷氨酰-l-缬氨酰-甘氨酸的方法线性范围为0.001~0.04μg/ml,线性相关系数为0.9995;检出限为0.3ng/ml,定量限为0.9ng/ml;日内和日间变异系数分别小于10%(n=5);以6种水产样品为例,其平均加标回收率在83.05%~102.19%之间,rsd为6.47%~9.30%。
附图说明
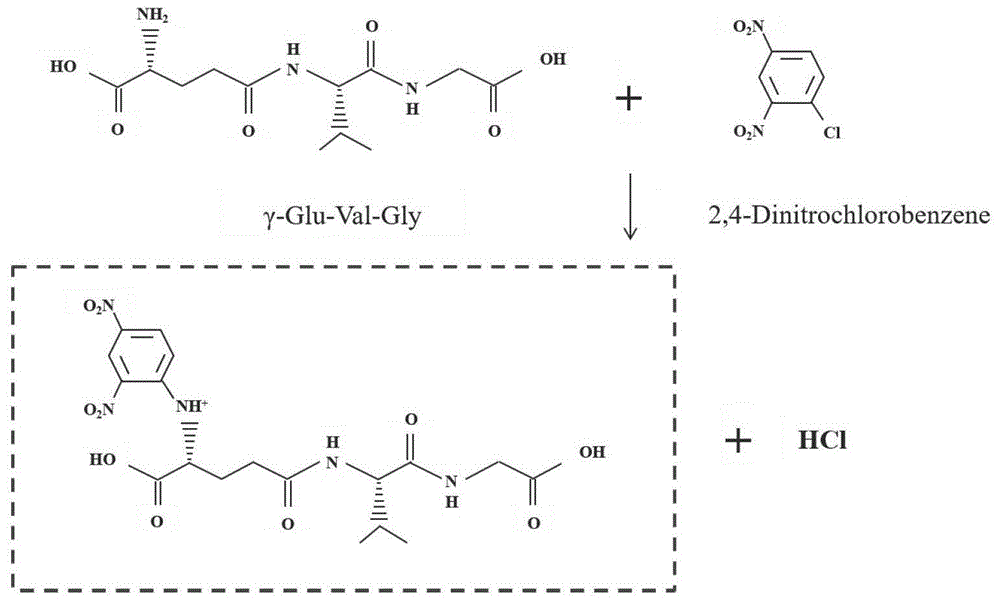
图1为本发明中γ-glu-val-gly与2,4-二硝基氯苯进行衍生化反应的示意图;
图2为γ-glu-val-gly标准品的衍生化产物(cdnb-evg)全扫描色谱图和质谱图;图2中(a)为全扫描色谱图,(b)为质谱图;
图3为γ-glu-val-gly标准品的衍生化产物(cdnb-evg)的定性、定量子离子质谱图;图3中(a)为定量子离子质谱图,(b)为定性子离子质谱图;
图4为内标物l-精氨酸-15n4标准品的衍生化产物(cdnb-arg)的全扫描色谱图和质谱图;图4中(a)为全扫描色谱图,(b)为质谱图;
图5为内标物l-精氨酸-15n4标准品的衍生化产物(cdnb-arg)的定量子离子质谱图;
图6为菲律宾帘蛤衍生化产物在多反应监测模式下的总离子流色谱图;图6中,(a)对应定量离子470.1/72.2色谱图,(b)对应定性离子470.1/395色谱图,(c)对应定性离子470.1/367色谱图,(d)对应定性离子470.1/184.2色谱图;
图7为kuroda团队文献报道中以aqc为衍生试剂的样品中γ-glu-val-gly衍生物色谱图,图7中(a)对应定量离子474.2/171.2色谱图,(b)对应定性离子474.2/145.3色谱图,(c)对应定性离子474.2/300.3色谱图,(d)对应定性离子474.2/229.4色谱图,(e)对应定性离子474.2/304.0色谱图,(f)对应定性离子474.2/72.1色谱图。
具体实施方式
本发明提供了一种检测水产品中呈味肽l-γ-谷氨酰-l-缬氨酰-甘氨酸的方法,包括以下步骤:
(1)提供待测水产品提取液,将所述待测水产品提取液与内标物溶液和去离子水混合,得到待测混合液;
(2)将所述待测混合液与缓冲溶液、2,4-二硝基氯苯溶液混合进行涡旋,得到待测涡旋液;将所述待测涡旋液在50~90℃条件下避光进行衍生化反应,得到待测进样液;
(3)将所述待测进样液进行液相色谱-串联质谱检测,得到γ-glu-val-gly色谱图和内标物色谱图;
(4)将由所述γ-glu-val-gly色谱图和内标物色谱图得到的γ-glu-val-gly和内标物的峰面积之比代入标准曲线,计算得到水产品中γ-glu-val-gly的含量;所述标准曲线为γ-glu-val-gly标准品和内标物的峰面积之比与γ-glu-val-gly标准品溶液浓度之间的线性曲线。
本发明提供待测水产品提取液。本发明对所述水产品没有特别的要求,本领域技术人员熟知的水产品均适用于本发明。在本发明中,所述提供待测水产品提取液的方法优选包括:
将待测水产品的可食部位剪碎后进行研磨,得到匀浆样品;
将所述匀浆样品与去离子水混合,将所得混合料液依次进行均质、搅拌和离心,得到上清液;
将所述上清液经0.45μm滤膜过滤,再将滤液进行超滤离心,所得下层滤液为所述待测水产品提取液。
本发明对所述研磨的方法没有特别的要求,采用本领域技术人员熟知的研磨方法将待测水产品的可食部位研碎即可。在本发明中,所述匀浆样品与去离子水的用量比优选为1g:10ml。在本发明中,所述均质和搅拌均优选在冰浴条件下进行;所述均质的速度优选为9000rpm,时间优选为3min;所述搅拌的速度优选为300rpm,时间优选为20min;所述离心的温度优选为4℃,离心力优选为5000g,离心的时间优选为10~15min,更优选为10min;本发明通过所述离心得到上清液和下层沉淀物,其中上清液为水和水溶多肽提取物,下层沉淀物为固体残渣。
在本发明中,所述超滤离心优选在超滤管中进行;所述超滤离心的温度优选为4℃,离心力优选为5000g,离心的时间优选为10~15min,更优选为15min;所述超滤离心的滤膜截留分子量优选为10kd;本发明通过所述超滤离心截留分子量大于10kd的成分,γ-glu-val-gly通过滤膜进入下层溶液,形成γ-glu-val-gly提取液。
得到待测水产品提取液后,本发明将所述提取液与内标物溶液和去离子水混合,得到待测混合液。在本发明中,所述内标物优选为l-精氨酸-15n4;所述内标物溶液的浓度优选为0.2mg/l;所述待测水产品提取液、内标物溶液和去离子水的体积比优选为1:1~2:1,更优选为1:1:1。
得到待测混合液后,本发明将所述待测混合液与缓冲溶液、2,4-二硝基氯苯溶液混合进行涡旋,得到待测涡旋液。在本发明中,所述缓冲溶液优选为na2co3-nahco3溶液;所述缓冲溶液的ph值优选为9.0;所述缓冲溶液与待测混合液的体积比优选为1:1~2:1,更优选为2:1。在本发明中,所述2,4-二硝基氯苯溶液优选为2,4-二硝基氯苯的乙腈溶液;所述2,4-二硝基氯苯溶液的质量浓度优选为5~20%,更优选为10%;所述2,4-二硝基氯苯溶液与待测混合液的体积比优选为1:1~1:3,更优选为1:1。本发明对所述涡旋的方法没有特别的要求,采用本领域技术人员熟知的涡旋方法保证混合均匀即可。
得到待测涡旋液后,本发明将所述待测涡旋液在50~90℃条件下避光进行衍生化反应,得到待测进样液。在本发明中,所述衍生化反应的温度优选为80~90℃,所述衍生化反应的温度优选通过水浴加热的方法实现;所述衍生化反应的时间优选为60~100min,更优选为90min。在本发明中,所述衍生化反应后还优选加入乙酸终止反应;所述乙酸的质量浓度优选为10%,本发明对所述乙酸的加入量没有特别的要求,能够将所述衍生化反应终止即可。本发明采用2,4-二硝基氯苯为衍生试剂,γ-glu-val-gly与2,4-二硝基氯苯(cdnb)之间发生亲核芳香取代反应,亲核阴离子由γ-glu-val-gly碱基电离生成,它攻击2,4-二硝基氯苯中含氯原子的碳,使氯离子离开从而生成γ-glu-val-gly衍生产物;内标物l-精氨酸-15n4同样与2,4-二硝基氯苯发生亲核芳香取代反应,生成内标物衍生产物。在本发明中,γ-glu-val-gly与2,4-二硝基氯苯进行衍生化反应示意图如图1所示。
在本发明中,所述衍生化反应终止后,还优选将所得反应液与水混合定容,然后经0.45μm滤膜过滤,滤液为所述待测进样液;在本发明实施例中,所述定容的体积优选为1ml;所述定容后还优选将混合液进行涡旋至均匀。
得到待测进样液后,本发明将所述待测进样液进行液相色谱-串联质谱检测,得到γ-glu-val-gly色谱图和内标物色谱图。在本发明中,所述液相色谱-串联质谱检测优选采用超高效液相色谱-三重四极杆质谱仪进行检测。在本发明中,所述液相色谱-串联质谱检测中液相色谱条件优选包括:c18色谱柱:粒径3μm,尺寸规格2.1mmid×100mm;柱温:35℃;流动相:a相为25mm甲酸水溶液,ph值为6.0;b相为60%体积分数的乙腈水溶液;流动相梯度洗脱程序:0~6min,b相体积分数35%;6.1~12min,b相体积分数100%;12.1~20min,b相体积分数35%;进样量:2.0μl;流速:0.2ml/min。
在本发明中,所述液相色谱-串联质谱检测中质谱条件优选包括:电喷雾离子源为正离子扫描模式;离子源参数:雾化气温度350℃,雾化气流速为12l/min,雾化器压力为20psi,毛细管电压为4.0kv;一级质谱数据采集采用全扫描模式,扫描范围为m/z50~500;二级质谱数据采集采用多反应监测模式;γ-glu-val-gly的定性离子对(m/z)为470.1/395(10v)、470.1/367(10v)和470.1/184.2(22v)、定量离子对为470.1/72.2(30v),内标物定量离子对为345.0/71.1;γ-glu-val-gly的衍生化产物(cdnb-evg)的传输电压优选为100v,内标物的衍生化产物(cdnb-arg)最佳的传输电压为140v。
得到γ-glu-val-gly色谱图和内标物色谱图后,本发明将由所述γ-glu-val-gly色谱图和内标物色谱图得到的γ-glu-val-gly和内标物的峰面积之比代入标准曲线,计算得到水产品中γ-glu-val-gly的含量;所述标准曲线为γ-glu-val-gly标准品和内标物的峰面积之比与γ-glu-val-gly标准品溶液浓度之间的线性曲线。在本发明中,所述计算的具体方法为:通过液相色谱-串联质谱检测得到待测水产品提取液中γ-glu-val-gly色谱图,以及对应的内标物溶液中内标物色谱图,将所述γ-glu-val-gly峰面积和内标物峰面积之比代入标准曲线中,通过计算得到提取液中γ-glu-val-gly浓度,再通过折算得到待测水产品中γ-glu-val-gly的含量。
在本发明中,所述标准曲线的获取方法优选包括以下步骤:
(a)分别将系列浓度的γ-glu-val-gly标准品溶液与内标物溶液和去离子水混合,得到系列浓度的标准混合液;
(b)分别将所述系列浓度的标准混合液与缓冲溶液和2,4-二硝基氯苯溶液混合进行涡旋,得到系列浓度的标准涡旋液;将所述标准涡旋液在50~90℃条件下避光进行衍生化反应,得到系列浓度的标准进样液;
(c)将所述标准进样液分别进行液相色谱-串联质谱检测,得到γ-glu-val-gly标准品色谱图和内标物色谱图,以γ-glu-val-gly标准品溶液的浓度为横坐标,以γ-glu-val-gly标准品与内标物对应的峰面积比为纵坐标,绘制得到标准曲线。
在本发明实施例中,所述γ-glu-val-gly标准品溶液的浓度分别为0.1μg/ml、0.2μg/ml、0.4μg/ml、0.6μg/ml和0.8μg/ml。在本发明中,所述步骤(a)~(c)的条件及操作与上述步骤(1)~(3)相同,在此不再赘述。
本发明采用的2,4-二硝基氯苯衍生试剂不仅稳定性好,而且价格低廉,以所述衍生试剂进行呈味肽l-γ-谷氨酰-l-缬氨酰-甘氨酸的检测,具有准确度高、灵敏度高、成本低的优点,且多反应监测模式下获得目标物定性、定量离子色谱图简单,无衍生副产物的干扰,可以满足水产品中呈味多肽l-γ-谷氨酰-l-缬氨酰-甘氨酸的定性、定量检测需求,并且操作简便。
下面结合实施例对本发明提供的检测水产品中呈味肽l-γ-谷氨酰-l-缬氨酰-甘氨酸的方法进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
实施例1
(1)标准品溶液的配制:分别配制浓度分别为0.1、0.2、0.4、0.6和0.8μg/ml的γ-glu-val-gly标准品水溶液。
(2)对水产样品(菲律宾帘蛤)进行前处理,获得样品提取液:
将菲律宾帘蛤的可食部位剪碎并研磨均匀,取10.00g样品,加入100ml去离子水,冰浴条件下9000rpm均质3min,300rpm磁力搅拌20min;取该溶液在4℃、5000g离心力的作用下离心10min,取上清液用0.45μm滤膜过滤;将滤液转移到超滤管(截留分子量大小为10kd)中,在4℃、5000g离心力的作用下超滤离心15min,收集下层滤液,获得菲律宾帘蛤样品γ-glu-val-gly提取液。
(3)衍生化反应:
取100μl待测混合液、200μl缓冲溶液(na2co3-nahco3,ph值为9.0)、100μl衍生化试剂溶液(10wt%2,4-二硝基氯苯/乙腈溶液)于1.5ml样品瓶中,充分涡旋后,90℃恒温水浴避光反应90min,反应结束后加入10wt%乙酸50μl终止反应,然后加水定容至1ml,涡旋混匀,经0.45μm滤膜过滤得待测溶液,准备上机检测。
其中,待测混合液分别从混合溶液①和混合溶液②中取出100μl,混合溶液①为:100μl标准品溶液+100μl内标物溶液+100μl去离子水;混合溶液②为:100μl菲律宾帘蛤样品提取液+100μl内标物溶液+100μl去离子水。内标物溶液为l-精氨酸-15n4溶液,浓度为0.2mg/l。
(4)将步骤(3)中得到的标准品待测溶液和菲律宾帘蛤待测溶液进行超高效液相色谱-三重四极杆质谱法检测,得到色谱图,内标法定量:
色谱条件:c18色谱柱(dikmainspire3μm,2.1mmid×100mm);柱温:35℃;流动相:a相为25mm甲酸水溶液(氨水调节ph值至6.0),b相为60%体积分数的乙腈水溶液;流动相梯度洗脱程序为:0~6min(b相体积分数35%),6.1~12min(b相体积分数100%),12.1~20min(b相体积分数35%);进样量:2.0μl;流速:0.2ml/min;
质谱条件:采用电喷雾离子源正离子扫描模式;离子源参数:雾化气温度350℃,雾化气流速为12l/min,雾化器压力为20psi,毛细管电压为4.0kv;一级质谱数据采集采用全扫描模式,扫描范围为m/z50~500,二级质谱数据采集采用多反应监测(mrm)模式;γ-glu-val-gly的衍生化产物(cdnb-evg)最佳的传输电压为100v,其定量子离子对为m/z470.1/72.2(30v)、定性子离子对为470.1/395(10v)、470.1/367(10v)、470.1/184.2(22v),内标物的衍生化产物(cdnb-arg)最佳的传输电压为140v,其定量子离子对为345/71.1(40v)。
图2为γ-glu-val-gly标准品的衍生化产物(cdnb-evg)全扫描色谱图(a)和质谱图(b);图3为γ-glu-val-gly标准品的衍生化产物(cdnb-evg)的定量子离子质谱图(a)、定性子离子质谱图(b);图4为内标物l-精氨酸-15n4标准品的衍生化产物(cdnb-arg)的全扫描色谱图(a)和质谱图(b);图5为内标物l-精氨酸-15n4标准品的衍生化产物(cdnb-arg)的定量子离子质谱图;图6为菲律宾帘蛤衍生化产物在多反应监测模式下的总离子流色谱图,与kuroda团队报道的结果(图7)相比,色谱图简单,只有γ-glu-val-gly、内标物的衍生物色谱峰,无衍生反应副产物干扰。
γ-glu-val-gly标准品和内标物经衍生后进行质谱分析,在全扫描(ms2scan)模式下,分别获得γ-glu-val-gly标准品和内标物衍生物的色谱图(图2a,图4a),确定二者的保留时间分别为1.861min和2.767min;根据对应的质谱图(图2b,图4b)分别确定γ-glu-val-gly标准品和内标物衍生物的母离子质荷比为470.1和345.0;在子离子扫描(productionscan)模式下,分别获得γ-glu-val-gly衍生物母离子(m/z470.1)和内标物衍生物母离子(m/z345)的二级离子质谱图(图3,图5),确定γ-glu-val-gly衍生物的定性和定量子离子的质荷比为72.2、395、367和184.2以及内标物衍生物定量子离子的质荷比为71.1;菲律宾帘蛤样品中γ-glu-val-gly经衍生后进行液质分析,在多反应监测(mrm)模式下,获得γ-glu-val-gly和内标物衍生物的定性、定量离子色谱图(图6),并将定量离子470.1/72.2(a)色谱图峰面积用于菲律宾帘蛤样品中γ-glu-val-gly定量分析。
内标法定量即以γ-glu-val-gly标准品溶液的浓度为横坐标,以γ-glu-val-gly标准品与内标物对应的峰面积比为纵坐标,绘制标准曲线,得到的标准曲线为y=0.4369x+0.0332,r2=0.9995。检出限和定量限的界定分别为3倍和10倍信噪比所对应的样品的浓度。该方法的线性范围为0.001~0.04μg/ml,线性相关系数为0.9995;检出限为0.3ng/ml,定量限为0.9ng/ml。
根据菲律宾帘蛤待测溶液的检测结果,对照标准曲线,获得菲律宾帘蛤待测溶液中γ-glu-val-gly的浓度,从而确定菲律宾帘蛤样品中γ-glu-val-gly的含量,菲律宾帘蛤样品中γ-glu-val-gly的含量为0.21±0.03mg/kg。
将0.1μg/ml的γ-glu-val-gly标准品溶液100μl加入到菲律宾帘蛤样品提取液中,进行加标回收率测定,加标回收率为83.05%,rsd为9.30%,重现性较好。衍生后的菲律宾帘蛤待测溶液在室温放置0、1、2、3、4d后进行测定,日内和日间变异系数小于10%(n=5)。
实施例2
检测海湾扇贝中呈味肽l-γ-谷氨酰-l-缬氨酰-甘氨酸的方法,步骤如下:
(1)样品提取:取海湾扇贝可食部分剪碎并研磨均匀,称取10.00g匀浆样品,加入100ml去离子水,冰浴条件下9000rpm均质3min,磁力搅拌20min,然后在4℃、5000g离心10min,取上清液过0.45μm滤膜,将滤液转移到超滤管(截留分子量大小为10kd)中,在4℃、5000g离心力作用下离心15min,收集下层滤液,获得待测样品提取液。
(2)衍生化反应:取100μl待测样品提取液、100μl内标物溶液(l-精氨酸-15n4溶液,浓度0.2mg/l)和100μl去离子水,涡旋混匀,得到待测混合液。取100μl待测混合液、200μl缓冲溶液(na2co3-nahco3,ph值9.0)、100μl衍生化试剂(10wt%2,4-二硝基氯苯/乙腈溶液)于1.5ml样品瓶中,充分涡旋后,90℃恒温水浴避光反应90min,然后加入10wt%乙酸50μl终止反应,加水定容至1ml,涡旋混匀,经0.45μm滤膜过滤得待测溶液,用超高效液相色谱-三重四极杆质谱仪进行定性、定量检测。
(3)定性、定量分析:
液相色谱条件:c18色谱柱(3μm,2.1mmid×100mm);柱温:35℃;流动相:a相25mm甲酸水溶液(氨水调节至ph6.0),b相60%体积分数的乙腈水溶液;流动相梯度洗脱程序为:0~6min(b相体积分数35%),6.1~12min(b相体积分数100%),12.1~20min(b相体积分数35%);进样量:2.0μl;流速:0.2ml/min;
质谱条件:电喷雾离子源正离子扫描模式;离子源参数:雾化气温度350℃,雾化气流速为12l/min,雾化器压力为20psi,毛细管电压为4.0kv;一级质谱数据采集采用全扫描模式,扫描范围为m/z50~500,二级质谱数据采集采用多反应监测(mrm)模式;γ-glu-val-gly的衍生化产物最佳的传输电压为100v。以离子对470.1/395(10v)、470.1/367(10v)、470.1/184.2(22v)定性γ-glu-val-gly;γ-glu-val-gly定量离子对为m/z470.1/72.2(30v),内标物定量离子对为345.0/71.1(40v)。
将检测结果,对照实施例1中标准曲线,获得待测溶液中γ-glu-val-gly的浓度,从而确定水产品样品中γ-glu-val-gly的含量。海湾扇贝样品中γ-glu-val-gly的含量为0.16±0.02mg/kg。
将0.1μg/ml的γ-glu-val-gly标准品溶液100μl加入到海湾扇贝样品提取液中,进行加标回收率测定,加标回收率为102.19%,rsd为8.61%。
实施例3
检测长牡蛎中呈味肽l-γ-谷氨酰-l-缬氨酰-甘氨酸的方法,步骤如下:
(1)样品提取:取长牡蛎可食部分剪碎并研磨均匀,称取10.00g匀浆样品,加入100ml去离子水,冰浴条件下9000rpm均质3min,磁力搅拌20min,然后在4℃、5000g离心力的作用下离心10min,取上清液过0.45μm滤膜,将滤液转移到超滤管(截留分子量大小为10kd)中,在4℃、5000g离心力的作用下超滤离心15min,收集下层滤液,获得待测样品提取液。
(2)衍生化反应:取100μl待测样品提取液、100μl内标物溶液(l-精氨酸-15n4溶液,浓度0.2mg/l)和100μl去离子水,涡旋混匀,得到待测混合液。取100μl待测混合液、200μl缓冲溶液(na2co3-nahco3,ph值9.0)、100μl衍生化试剂(10wt%2,4-二硝基氯苯/乙腈溶液)于1.5ml样品瓶中,充分涡旋后,90℃恒温水浴避光反应90min,然后加入10wt%乙酸50μl终止反应,加水定容至1ml,涡旋混匀,经0.45μm滤膜过滤得待测溶液,用超高效液相色谱-三重四极杆质谱仪进行定性、定量检测。
(3)定性、定量分析:
液相色谱条件:c18色谱柱(3μm,2.1mmid×100mm);柱温:35℃;流动相:a相25mm甲酸水溶液(氨水调节ph值至6.0),b相60%体积分数的乙腈水溶液;流动相梯度洗脱程序为:0~6min(b相体积分数35%),6.1~12min(b相体积分数100%),12.1~20min(b相体积分数35%);进样量:2.0μl;流速:0.2ml/min;
质谱条件:电喷雾离子源正离子扫描模式;离子源参数:雾化气温度350℃,雾化气流速为12l/min,雾化器压力为20psi,毛细管电压为4.0kv;一级质谱数据采集采用全扫描模式,扫描范围为m/z50-500,二级质谱数据采集采用多反应监测(mrm)模式;γ-glu-val-gly的衍生化产物最佳的传输电压为100v;以离子对470.1/395(10v)、470.1/367(10v)、470.1/184.2(22v)定性γ-glu-val-gly;γ-glu-val-gly定量离子对为m/z470.1/72.2(30v),内标物定量离子对为345.0/71.1(40v)。
将检测结果,对照实施例1中标准曲线,获得待测溶液中γ-glu-val-gly的浓度,从而确定水产品样品中γ-glu-val-gly的含量。长牡蛎样品中γ-glu-val-gly的含量为0.27±0.09mg/kg。
将0.1μg/ml的γ-glu-val-gly标准品溶液100μl加入到长牡蛎样品提取液中,进行加标回收率测定,加标回收率为95.17%,rsd为9.04%。
实施例4
检测方形马珂蛤中呈味肽l-γ-谷氨酰-l-缬氨酰-甘氨酸的方法,步骤如下:
(1)样品提取:取方形马珂蛤可食部分剪碎并研磨均匀,称取10.00g匀浆样品,加入100ml去离子水,冰浴条件下9000rpm均质3min,磁力搅拌20min,然后在4℃、5000g离心力的作用下离心10min,取上清液过0.45μm滤膜,将滤液转移到超滤管(截留分子量大小为10kd)中,在4℃、5000g离心力的作用下超滤离心15min,收集下层滤液,获得待测样品提取液。
(2)衍生化反应:取100μl待测样品提取液、100μl内标物溶液(l-精氨酸-15n4溶液,浓度0.2mg/l)和100μl去离子水,涡旋混匀,得到待测混合液。取100μl待测混合液、200μl缓冲溶液(na2co3-nahco3,ph值9.0)、100μl衍生化试剂(10wt%2,4-二硝基氯苯/乙腈溶液)于1.5ml样品瓶中,充分涡旋后,90℃恒温水浴避光反应90min,然后加入10wt%乙酸50μl终止反应,加水定容至1ml,涡旋混匀,经0.45μm滤膜过滤得待测溶液,用超高效液相色谱-三重四极杆质谱仪进行定性、定量检测。
(3)定性、定量分析:
液相色谱条件:c18色谱柱(3μm,2.1mmid×100mm);柱温:35℃;流动相:a相25mm甲酸水溶液(氨水调节至ph6.0),b相60%体积分数的乙腈水溶液;流动相梯度洗脱程序为:0~6min(b相体积分数35%),6.1~12min(b相体积分数100%),12.1~20min(b相体积分数35%);进样量:2.0μl;流速:0.2ml/min;
质谱条件:电喷雾离子源正离子扫描模式;离子源参数:雾化气温度350℃,雾化气流速为12l/min,雾化器压力为20psi,毛细管电压为4.0kv;一级质谱数据采集采用全扫描模式,扫描范围为m/z50-500,二级质谱数据采集采用多反应监测(mrm)模式;γ-glu-val-gly的衍生化产物最佳的传输电压为100v;以离子对470.1/395(10v)、470.1/367(10v)、470.1/184.2(22v)定性γ-glu-val-gly;γ-glu-val-gly定量离子对为m/z470.1/72.2(30v),内标物定量离子对为345.0/71.1(40v)。
将检测结果,对照实施例1中标准曲线,获得待测溶液中γ-glu-val-gly的浓度,从而确定水产品样品中γ-glu-val-gly的含量。方形马珂蛤样品中γ-glu-val-gly的含量为0.18±0.02mg/kg。
将0.1μg/ml的γ-glu-val-gly标准品溶液100μl加入到方形马珂蛤样品提取液中,进行加标回收率测定,加标回收率为99.24%,rsd为8.09%。
实施例5
检测皱纹盘鲍中呈味肽l-γ-谷氨酰-l-缬氨酰-甘氨酸的方法,步骤如下:
(1)样品提取:取皱纹盘鲍可食部分剪碎并研磨均匀,称取10.00g匀浆样品,加入100ml去离子水,冰浴条件下9000rpm均质3min,磁力搅拌20min,然后在4℃、5000g离心10min,取上清液过0.45μm滤膜,将滤液转移到超滤管(截留分子量大小为10kd)中,在4℃、5000g离心力的作用下离心15min,收集下层滤液,获得待测样品提取液。
(2)衍生化反应:取100μl待测样品提取液、100μl内标物溶液(l-精氨酸-15n4溶液,浓度0.2mg/l)和100μl去离子水,涡旋混匀,得到待测混合液。取100μl待测混合液、200μl缓冲溶液(na2co3-nahco3,ph值9.0)、100μl衍生化试剂(10wt%2,4-二硝基氯苯/乙腈溶液)于1.5ml样品瓶中,充分涡旋后,90℃恒温水浴避光反应90min,然后加入10wt%乙酸50μl终止反应,加水定容至1ml,涡旋混匀,经0.45μm滤膜过滤得待测溶液,用超高效液相色谱-三重四极杆质谱仪进行定性、定量检测。
(3)定性、定量分析:
液相色谱条件:c18色谱柱(3μm,2.1mmid×100mm);柱温:35℃;流动相:a相25mm甲酸水溶液(氨水调节至ph6.0),b相60%体积分数的乙腈水溶液;流动相梯度洗脱程序为:0~6min(b相体积分数35%),6.1~12min(b相体积分数100%),12.1~20min(b相体积分数35%);进样量:2.0μl;流速:0.2ml/min;
质谱条件:电喷雾离子源正离子扫描模式;离子源参数:雾化气温度350℃,雾化气流速为12l/min,雾化器压力为20psi,毛细管电压为4.0kv;一级质谱数据采集采用全扫描模式,扫描范围为m/z50~500,二级质谱数据采集采用多反应监测(mrm)模式;γ-glu-val-gly的衍生化产物最佳的传输电压为100v;以离子对470.1/395(10v)、470.1/367(10v)、470.1/184.2(22v)定性γ-glu-val-gly;γ-glu-val-gly定量离子对为m/z470.1/72.2(30v),内标物定量离子对为345.0/71.1(40v)。
将检测结果,对照实施例1中标准曲线,获得待测溶液中γ-glu-val-gly的浓度,从而确定水产品样品中γ-glu-val-gly的含量。皱纹盘鲍样品中γ-glu-val-gly的含量为0.25±0.03mg/kg。
将0.1μg/ml的γ-glu-val-gly标准品溶液100μl加入到皱纹盘鲍样品提取液中,进行加标回收率测定,加标回收率为93.46%,rsd为6.47%。
实施例6
检测南美白对虾中呈味肽l-γ-谷氨酰-l-缬氨酰-甘氨酸的方法,步骤如下:
(1)样品提取:取南美白对虾可食部分剪碎并研磨均匀,称取10.00g匀浆样品,加入100ml去离子水,冰浴条件下9000rpm均质3min,磁力搅拌20min,然后在4℃、5000g离心力的作用下离心10min,取上清液过0.45μm滤膜,将滤液转移到超滤管(截留分子量大小为10kd)中,在4℃、5000g离心力的作用下超滤离心15min,收集下层滤液,获得待测样品提取液。
(2)衍生化反应:取100μl待测样品提取液、100μl内标物溶液(l-精氨酸-15n4溶液,浓度0.2mg/l)和100μl去离子水,涡旋混匀,得到待测混合液。取100μl待测混合液、200μl缓冲溶液(na2co3-nahco3,ph值9.0)、100μl衍生化试剂(10wt%2,4-二硝基氯苯/乙腈溶液)于1.5ml样品瓶中,充分涡旋后,90℃恒温水浴避光反应90min,然后加入10wt%乙酸50μl终止反应,加水定容至1ml,涡旋混匀,经0.45μm滤膜过滤得待测溶液,用超高效液相色谱-三重四极杆质谱仪进行定性、定量检测。
(3)定性、定量分析:
液相色谱条件:c18色谱柱(3μm,2.1mmid×100mm);柱温:35℃;流动相:a相25mm甲酸水溶液(氨水调节ph值至6.0),b相60%体积分数的乙腈水溶液;流动相梯度洗脱程序为:0~6min(b相体积分数35%),6.1~12min(b相体积分数100%),12.1~20min(b相体积分数35%);进样量:2.0μl;流速:0.2ml/min;
质谱条件:电喷雾离子源正离子扫描模式;离子源参数:雾化气温度350℃,雾化气流速为12l/min,雾化器压力为20psi,毛细管电压为4.0kv;一级质谱数据采集采用全扫描模式,扫描范围为m/z50-500,二级质谱数据采集采用多反应监测(mrm)模式;γ-glu-val-gly的衍生化产物最佳的传输电压为100v;以离子对470.1/395(10v)、470.1/367(10v)、470.1/184.2(22v)定性γ-glu-val-gly;γ-glu-val-gly定量离子对为m/z470.1/72.2(30v),内标物定量离子对为345.0/71.1(40v)。
将检测结果,对照实施例1中标准曲线,获得待测溶液中γ-glu-val-gly的浓度,从而确定水产品样品中γ-glu-val-gly的含量。南美白对虾样品中γ-glu-val-gly的含量为0.24±0.01mg/kg。
将0.1μg/ml的γ-glu-val-gly标准品溶液100μl加入到南美白对虾样品提取液中,进行加标回收率测定,加标回收率为97.83%,rsd为7.53%。
由以上实施例可以看出,本发明以2,4-二硝基氯苯为衍生化试剂,采用液相色谱-串联质谱内标法测定水产品中γ-glu-val-gly含量,操作简便易行、方法准确度高,衍生剂价廉易得,可以用于水产品中γ-glu-val-gly的定性和定量分析。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!