一种沙参麦冬汤的HPLC指纹图谱的构建方法与流程
一种沙参麦冬汤的hplc指纹图谱的构建方法
技术领域
[0001]
本发明属于一种医药技术领域,具体是涉及到一种沙参麦冬汤的hplc指纹图谱的构建方法。
背景技术:
[0002]
经典名方是指目前广泛应用、疗效确切、具有明显特色与优势的清代及清代以前医药典籍中所记载的方剂。2018年4月16日,国家中医药管理局颁布了包括沙参麦冬汤在内的100首古代经典名方,沙参麦冬汤为第75方。经典名方多为复方,由多种药味组成,所含化学成分复杂,仅靠其中几个成分的含量来控制整体质量显然不合理。
[0003]
沙参麦冬汤源于温病学代表著作《温病条辨》,是清代名医吴鞠通为治疗温病后期燥伤肺胃阴液而创立。该方由北沙参、麦冬、甘草、桑叶、玉竹、白扁豆、天花粉组成,其中北沙参和麦冬为君药,玉竹和天花粉为臣药,白扁豆和桑叶为佐药,甘草为使药,具有清养肺胃、生津润燥的作用,是治疗肺胃阴伤的名方。目前,关于沙参麦冬汤的研究多集中在药理作用及临床应用等方面,现代药理学研究表明,沙参麦冬汤有抗炎、抗肿瘤、调节免疫功能的作用,临床常用于治疗呼吸系统疾病,疗效显著,是清养肺胃,生津润燥的代表方剂,为清金保肺的要药,而有关沙参麦冬汤的指纹图谱研究报道较少。
技术实现要素:
[0004]
本发明要解决的技术问题是提供一种沙参麦冬汤的hplc指纹图谱的构建方法,hplc指纹图谱结合聚类分析与主成分分析同时测定的方法,快速、简便、重复性好,能够为沙参麦冬汤及其制剂的质量评价提供参考。
[0005]
本发明的内容包括沙参麦冬汤的hplc指纹图谱的构建方法,包括如下步骤:
[0006]
1)供试品溶液的制备:称取沙参麦冬汤药材,加水1000ml,煎煮浓缩至400ml,过滤,得沙参麦冬汤,移取4ml沙参麦冬汤至10ml容量瓶,加甲醇定容,得供试品溶液;
[0007]
2)对照品溶液的制备:称取芦丁、甘草苷、甘草酸铵对照品适量,加甲醇配制成芦丁的质量浓度为0.0733mg/ml、甘草苷的质量浓度为0.04mg/ml、甘草酸铵的质量浓度为0.0667mg/ml的混合对照品溶液;
[0008]
3)高效液相色谱检测:
[0009]
色谱条件为:色谱柱为c
18
色谱柱,柱温为30℃,采用流动相a为乙腈,流动相b为0.2%体积浓度的磷酸水溶液,流速为1ml/min;进行梯度洗脱,梯度洗脱程度为:
[0010]
0~5min:5~15%乙腈,5~15min:15~23%乙腈,15~20min:23~35%乙腈,20~25min:35~45%乙腈,25~40min:45~55%乙腈,40~50min:55~60%乙腈,50~60min:60~80%乙腈,60~70min:80~100%乙腈;
[0011]
检测波长程序为0~5min:354nm,5.01~30min:230nm,30.01~40min:203nm,41.01~50min:230nm,50.01~70min:203nm;
[0012]
4)沙参麦冬汤的hplc指纹图谱的构建:吸取供试品溶液、对照品溶液各20μl,注入
高效液相色谱仪中,用步骤3)的高效液相色谱检测,匹配26个特征峰,确定3个共有峰,建立沙参麦冬汤的hplc指纹图谱。
[0013]
沙参麦冬汤的具体制备方法为:称取沙参、麦冬各11.19g,冬桑叶、天花粉、白扁豆各5.60g,玉竹7.46g,生甘草3.73g,加水1000ml,煎煮至剩余400ml,过滤,得沙参麦冬汤。
[0014]
所述色谱柱为zorbax c18,尺寸为250mm
×
4.6mm.,5.0μm。
[0015]
本发明的有益效果是,本申请建立了15批沙参麦冬汤的hplc指纹图谱,相似度均>0.90,标定了26个共有峰,指认出3个色谱峰,分别为13号峰(芦丁)、15号峰(甘草苷)及26号峰(甘草酸铵),聚类分析将15批次聚为3类,主成分分析将15批次聚为2类。本申请的hplc指纹图谱,结合聚类分析与主成分分析同时测定的方法,快速、简便、重复性好,客观反映沙参麦冬汤化学成分的整体性、复杂性及其质量差异,以期为后续沙参麦冬汤的物质基准研究提供前期基础,并为其他经典名方的研究提供参考。
附图说明
[0016]
图1为空白甲醇的指纹图谱。
[0017]
图2为混合对照品的指纹图谱,其中13为芦丁,15为甘草苷、26为甘草酸铵。
[0018]
图3为沙参麦冬汤指纹图谱。
[0019]
图4为15批次沙参麦冬汤指纹图谱。
[0020]
图5为单味药材及缺单味药材阴性供试品指纹图谱。
[0021]
图6为15批次沙参麦冬汤指纹图谱的聚类分析。
[0022]
图7为主成分分析的碎石图。
[0023]
图8为对比文献方法的15批次沙参麦冬汤指纹图谱(注:8.甘草苷;17.甘草酸铵)。
具体实施方式
[0024]
实施例1
[0025]
1.仪器与材料
[0026]
1.1仪器
[0027]
安捷伦1200高效液相色谱仪(四元泵,自动进样器及紫外检测器),美国安捷伦科技有限公司;sb-5200d型超声波清洗机,宁波新芝生物科技股份有限公司;xb 220a型电子天平(d=0.01g),广州广电计量检测股份有限公司。
[0028]
1.2试剂与试药
[0029]
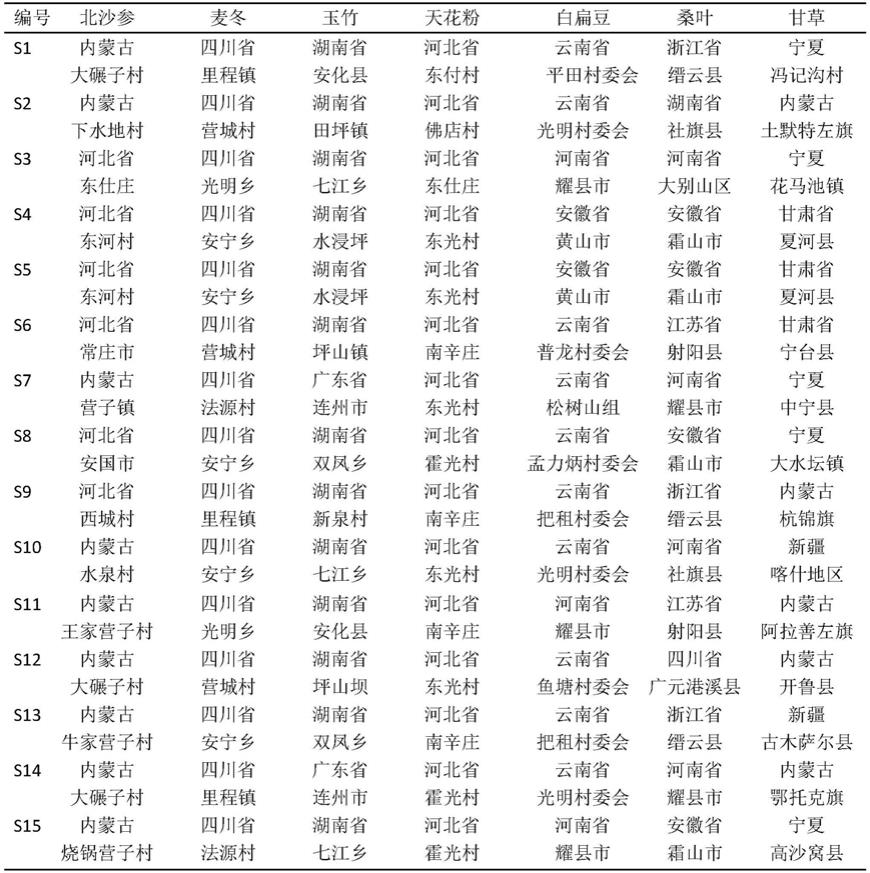
芦丁对照品(批号100080-201811)、甘草苷对照品(批号111610-201908)及甘草酸铵对照品(批号110731-202021),中国食品药品检定研究院;甲醇、乙腈均为色谱纯(斯百全化学有限公司);磷酸为分析纯(国药集团化学试剂有限公司);水为纯化水。15批沙参麦冬汤药材饮片来源见表1,经湖南中医药大学中药鉴定教研室主任刘塔斯教授鉴定,均符合2020年版《中国药典》标准。
[0030]
表1 15批次沙参麦冬汤药材饮片来源
[0031][0032]
2.方法与结果
[0033]
2.1煎煮方法
[0034]
《温病条辩》记载原方用法用量为“沙参三钱,玉竹二钱,生甘草一钱,冬桑叶一钱五分,麦冬三钱,生扁豆一钱五分,花粉一钱五分,以水五杯,煮取两杯,日再服”,据清朝度量衡的考证,1两=10钱=100分=37.3g,一杯之容量约为200ml。最终确定沙参麦冬汤的煎煮方法为:称取沙参、麦冬各11.19g,冬桑叶、天花粉、白扁豆各5.60g,玉竹7.46g,生甘草3.73g,加水1000ml,煎煮至剩余400ml,滤过即得沙参麦冬汤煎液。
[0035]
2.2色谱条件
[0036]
色谱柱为zorbax c18(250mm
×
4.6mm.5.0μm),流动相为乙腈-0.2%磷酸水,柱温为30℃,体积流量为1.0ml/min,进样量为20μl,梯度洗脱程序为0~5min:5~15%乙腈,5~15min:15~23%乙腈,15~20min:23~35%乙腈,20~25min:35~45%乙腈,25~40min:45~55%乙腈,40~50min:55~60%乙腈,50~60min:60~80%乙腈,60~70min:80~100%乙腈,检测波长程序为0~5min:354nm,5.01~30min:230nm,30.01~40min:203nm,41.01~50min:230nm,50.01~70min:203nm。
[0037]
2.3溶液的制备
[0038]
2.3.1混合对照品溶液的制备
[0039]
精密称取芦丁、甘草苷、甘草酸铵对照品适量,加甲醇配制成芦丁的质量浓度为0.0733mg/ml、甘草苷的质量浓度为0.04mg/ml、甘草酸铵的质量浓度为0.0667mg/ml的混合对照品溶液。
[0040]
2.3.2供试品溶液的制备
[0041]
称取处方量的沙参麦冬汤药材,加水1000ml,煎煮浓缩至400ml,滤过得沙参麦冬汤煎液,精密移取4ml沙参麦冬汤煎液至10ml容量瓶,加甲醇定容至刻度线,即得全方供试品溶液。单味药材供试品溶液及缺单味药材阴性供试品溶液均按上述方法进行制备。
[0042]
2.4沙参麦冬汤指纹图谱的方法学考察
[0043]
2.4.1精密度试验
[0044]
取同一供试品溶液(s1),在“2.2”项色谱条件下,连续进样6次,因甘草苷峰面积相对较大且稳定,故以其为参照峰,测得各共有峰相对保留时间和相对峰面积的rsd值均小于3%,相似度均大于0.9,表明该方法精密度良好。
[0045]
2.4.2重复性试验
[0046]
分别取供试品溶液(s1)6份,在“2.2”项色谱条件下进样检测,以甘草苷为参照峰,测得各共有峰相对保留时间和相对峰面积的rsd值均小于3%,相似度均大于0.9,表明该方法重复性良好。
[0047]
2.4.3稳定性试验
[0048]
取同一供试品溶液(s1),在“2.2”项色谱条件下,分别在0h、3h、6h、9h、12h及24h进样检测,以甘草苷为参照峰,测得各共有峰相对保留时间和相对峰面积的rsd值均小于3%,相似度均大于0.9,表明供试品溶液在24h内稳定。
[0049]
2.5沙参麦冬汤指纹图谱的建立及评价
[0050]
2.5.1指纹图谱的建立及相似度评价
[0051]
取15批沙参麦冬汤药材,按“2.3.2”项下供试品制备方法制备,在“2.2”项下色谱条件进样分析,记录沙参麦冬汤指纹图谱,并以甘草苷为参照峰,采用国家药典委员会发行的《中药色谱指纹图谱相似度评价系统2012a版》对15批沙参麦冬汤指纹图谱进行相似度分析,设定s1为参照图谱,利用中位数法,时间窗宽度设置0.1min,共标定26个共有峰,对照指纹图谱见图1-3,匹配后的15批次沙参麦冬汤指纹图谱见图4。
[0052]
经由《中药色谱指纹图谱相似度评价系统2012a版》计算,各批次与对照指纹图谱进行比较,相似度分别为:s1:0.985,s2:0.930,s3:0.994,s4:0,945,s5:0.91
[0053]
0,s6:0.922,s7:0.907,s8:0.944.s9:0.945,s10:0.908,s11:0.920,s12:0.917,s13:0.904,s14:0.916,s15:0.989。各批次样品相似度均在0.90-0.989之间,表明各批次沙参麦冬汤质量相对稳定,且由图1-4可知各产地沙参麦冬汤药材内在成分与对照指纹图谱基本相似,表明各化学成分基本一致,但部分峰的峰面积有所差异,表明部分产地所含化学成分的量有所不同,且根据对照品指认出3个共有峰,分别是13号峰(芦丁)、15号峰(甘草苷)及26号峰(甘草酸铵)。
[0054]
2.5.2指纹图谱共有峰的归属
[0055]
单味药材供试品溶液及缺单味药材阴性供试品溶液均按“2.3.2”项下供试品制备方法制备,在“2.2”项下色谱条件进样分析,记录色谱图,见图5。
[0056]
将色谱图与全方提取液指纹图谱比较进行单味药材的峰归属,结果如下:1号峰为除甘草外,其他药材的共有峰,2号峰为桑叶、北沙参及玉竹的共有峰,3号峰为白扁豆的特有峰,4号、10号、16号及17号峰为桑叶的特有峰,5号峰为北沙参、天花粉及玉竹的共有峰,6号峰为除天花粉及甘草外,其他药材的共有峰,7号、13号及18号峰为桑叶及甘草的共有峰,8号、14号、15号、19号、20号、21号、22号、23号、24号、25号及26号峰为甘草的特有峰,9号、11号及12号峰为所有药材的共有峰。
[0057]
2.5.3指纹图谱的聚类分析
[0058]
将各共有峰的相对峰面积导入spss 21.0统计软件,以甘草苷为参照峰,采用ward
’
s method方法,以欧氏距离(euclidean distance)为距离公式,对15批次沙参麦冬汤指纹图谱进行聚类分析,结果见图6。
[0059]
聚类分析结果显示,当距离为5时,可将15批次聚为三类,s1、s2、s3、s4、s8、s9、s15聚为一类,s5、s6、s11、s12、s14聚为一类,s7、s10、s13聚为一类,与相似度结果进行比较发现:s7、s10、s13相似度均在0.90-0.91之间,s5、s6、s11、s12、s14相似度均在0.91-0.93之间,s1、s2、s3、s8、s9、s15相似度均在0.93-0.99之间,聚类分析结果与相似度分析结果基本一致。总体看来,各产地沙参麦冬汤药材质量大体相近。
[0060]
2.5.4指纹图谱的主成分分析
[0061]
将各共有峰的峰面积导入spss 21.0统计软件,对15批次沙参麦冬汤的指纹图谱进行主成分分析,计算主成分特征值、累计贡献率并进行综合评分。
[0062]
相关系数的特征值及累计贡献率见表2,初始因子载荷矩阵见表3,碎石图见图5。表2中前7个主成分的累计方差贡献率为93.063%>85%,代表了沙参麦冬汤指纹图谱中26个共有峰93.063%的信息量,且由碎石图可发现前7个主成分之后曲线有一个明显的拐点,表明前7个主成分的分析结果基本显示出了不同批次沙参麦冬汤指纹图谱间的相似度与差异性,前7个主成分具有较强的代表性,因此选择前7个主成分进行评价。由表3可知第1主成分主要反映了14、20及26号峰的信息,第2主成分主要反映了4及16号峰的信息,第3主成分主要反映了13号峰的信息,第4主成分主要反映了2号峰的信息,第5主成分主要反映了24号峰的信息,第6主成分主要反映了3号峰的信息,第7主成分主要反映了8号峰的信息。这些峰较大程度地表征了沙参麦冬汤的整体信息,可选择这些特征峰作为沙参麦冬汤制剂的重要质控指标。
[0063]
表2特征值及累计贡献率
[0064][0065]
表3初始因子载荷矩阵
[0066][0067][0068]
根据初始因子载荷矩阵及特征值计算特征向量,进而计算得到7个主成分得分f1、f2、f3、f4、f5、f6及f7,由7个主成分的方差贡献率为权重系数,计算综合得分,f=0.32815*f1+0.19877*f2+0.1575*f3+0.09105*f4+0.06343*f5+0.04965*f6+0.04208*f7,结果见表4。从得分结果可看出,主成分分析将15批次分为两类,得分最低的s7、s10及s13为一类,其余为一类,这与聚类分析结果基本一致。
[0069]
表4主成分得分、综合得分及排名
[0070][0071]
对比例1
[0072]
参考文献(陈薛静,秦文杰,薛慧清,等.经典名方沙参麦冬汤特征图谱及含量测定研究[j].中国中医药信息杂志,2020,27(09):87-91。)方法,设计对比例1。
[0073]
不同方法的洗脱程序和检测波长如下表5所示。
[0074]
表5不同方法的洗脱程序和检测波长表
[0075][0076]
通过对比,本申请的洗脱程序和检测波长程序的间隔时间较文献更短,流动相比例变化更慢,洗脱出更多的峰。本法指纹图具有信息量大(共有峰多,26个)、分析时间短(30min)等特点,对比例1的文献方法共有峰20个,分析时间约70min。
[0077]
对比例1的指纹图谱如图8所示。
[0078]
对比结果:
[0079]
本申请的洗脱程序得到的特征图谱效果更好,效果有:本法指纹图具有信息量大(共有峰多:26个)、分析时间短(30min)、各批次样品相似度更接近(均在0.90-0.989之间)等特点。文献方法得到的信息量较少(共有峰少:20个)、分析时间长(约70min)、各批次样品相似度均在0.86-1.00之间。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1