一种青黛辐照前后主要成分的测定方法与流程
1.本发明属于中医药技术领域,具体涉及一种青黛辐照前后主要成分的测定方法。
背景技术:
2.青黛indigonaturalis,别名靛花、青蛤粉、青缸花、蓝露、淀花、靛沫花,质量标准收载于《中国药典》,来源为爵床科植物马蓝baphicacanthuscusia(nees)bremek.、蓼科植物蓼蓝polygonumtinctoriumait.、或十字花科植物菘蓝isatisindigoticafort.的叶或茎叶经加工制得的干燥粉末、团块或颗粒。具有清热解毒,凉血消斑,泻火定惊的功效。主治温毒发斑,血热吐衄,咽痛口疮,火毒疮疡,咳嗽胸痛,痰中带血,暑热惊痫,惊风抽搐。
3.主要化学成分:含靛蓝、靛玉红、靛棕、靛黄、鞣酸、β
‑
谷甾醇、蛋白质和大量无机盐。
4.药理作用:青黛对金黄色葡萄球菌、炭疽杆菌、志贺氏痢疾杆菌、霍乱弧菌均有抗抑作用。所含靛玉红为其抗癌有效成分,对移植性肿瘤有中等强度的抑制作用。
5.加工方法:采收落叶,加水浸泡,至叶腐烂,叶落脱皮时,捞去落叶,加适量石灰乳,充分搅拌至浸液由乌绿色转为深红色时,捞取液面泡沫,晒干而成。
6.青黛广泛用于中医临床治疗中,多入散剂和丸剂使用。由于中药材的特性,在采收加工及生产过程中,均可能含有大量的微生物,根据《中国药典》附录要求,散剂有微生物限度控制要求,因此,青黛用于散剂制剂,就需要对青黛中的微生物进行处理和研究。企业通常采用钴60辐照进行灭菌处理,但辐照灭菌应用于传统中药的灭菌历史尚短,基础研究尚不完善,根据国家食品药品监督管理局颁布的《中药辐照灭菌技术指导原则》,中药采用辐照灭菌应充分说明其必要性。青黛辐照前后的质量研究,对保证产品的质量和临床疗效有着重要意义。
技术实现要素:
7.本发明的目的在于提供一种青黛辐照前后主要成分的测定方法,对青黛辐照前后的质量进行研究,对保证产品的质量和临床疗效有着重要意义。
8.为了实现上述目的,本发明采用以下技术方案:
9.一种青黛辐照前后主要成分的测定方法,包括以下步骤:
10.(1)青黛辐照前后靛蓝和靛玉红含量的测定:
11.对照品溶液的制备:取靛蓝或靛玉红对照品2.5mg,精密称定,置250ml容量瓶中,加2%水合氯醛的三氯甲烷溶液约220ml,超声处理1.5小时,放冷,加2%水合氯醛的三氯甲烷溶液至刻度,摇匀,即得每l ml中含靛蓝或靛玉红10μg的靛蓝或靛玉红对照品溶液;
12.供试品溶液的制备:取青黛样品细粉50mg,精密称定,置250ml容量瓶中,加n,n
‑
二甲基甲酰胺约220ml,超声处理30分钟,放冷,加n,n
‑
二甲基甲酰胺至刻度,摇匀,滤过,取续滤液,即得;
13.样品分析:取3批次辐照前后样品s1~s3,分别平行制备对照品和供试品溶液各2
份,色谱条件下平行测定2次,记录峰面积,采用外标一点法计算样品中靛蓝和靛玉红含量;
14.(2)青黛辐照前后整体化学成分的测定:
15.对照品溶液的制备:分别取靛蓝、靛玉红对照品1.0mg,精密称定,加甲醇制成每1ml含0.1mg的溶液,即得;
16.供试品溶液的制备:取青黛粉末250mg,精密称定,置具塞锥形瓶中,精密加入甲醇25ml,称定重量,超声提取30min,放冷,再称定重量,称重后甲醇补足失重,摇匀滤过,取续滤液,即得;
17.指纹图谱测定:精密吸取对照品溶液和供试品溶液各20μl,注入高效液相色谱仪,测定,得到各批次样品色谱图和各色谱峰峰面积,比较同批次辐照后样品与未辐照样品之间的差异;
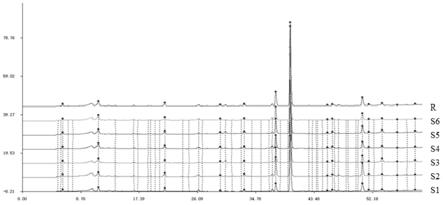
18.指纹图谱的建立:取3批次辐照前后青黛样品s1~s3共计6份样品,测定指纹图谱,将s1设为参照谱,时间宽度为0.10,采用中位数法,生成对照图谱,确定其中14个色谱峰为青黛的指纹图谱共有峰,建立指纹图谱,并以39.99min左右处出峰的靛玉红为参照峰s,取3批次青黛药材辐照前后样品和对照药材指纹图谱计算各色谱峰保留时间和保留峰面积与同一图谱中s峰的保留时间和保留峰面积的比值,得到相对保留时间和相对峰面积,比较特征峰即图谱的相似度;
19.(3)青黛辐照前后稳定性考察:
20.青黛辐照前后样品中靛蓝和靛玉红含量稳定性考察:取3批次辐照前后青黛样品s1~s3,放置0、6、7、8个月后,采用步骤(1)中青黛含量测定方法对辐照前后靛蓝、靛玉红含量(%)进行测定,以此判断辐照对青黛药材中靛蓝、靛玉红含量稳定性的影响;
21.青黛辐照前后样品中整体化学成分稳定性考察:取3批次辐照前后青黛样品s1~s3,放置0、6、7、8个月后,采用步骤(2)中青黛指纹图谱分析方法对辐照前后青黛样品进行指纹图谱分析,以此判断辐照对青黛样品整体化学成分稳定性的影响。
22.进一步的,所述青黛辐照前后靛蓝和靛玉红含量的测定中,三氯甲烷溶液的配置过程为:取水合氯醛,置硅胶干燥器中放置24小时,称取2.0g,加三氯甲烷至100ml,放置,出现浑浊,以无水硫酸钠脱水,滤过,即得。
23.进一步的,所述青黛辐照前后靛蓝和靛玉红含量的测定中,超声处理的功率均为250w,频率均为33khz。
24.进一步的,所述青黛辐照前后靛蓝和靛玉红含量的测定中,色谱条件为:以甲醇
‑
水(70:30)为流动相,检测波长为292nm,柱温25℃,进样量10μl。
25.进一步的,所述青黛辐照前后整体化学成分的测定中,指纹图谱测定中,色谱仪的检测波长为242nm;流动相为乙腈(b)
‑
0.02%磷酸水(ph=3)(a);流动相梯度为0~5min,20%b,5~35min,20%~55%b,35~45min,55%~66%b,45~60min,66%~95%b;流速为0.8ml/min。
26.进一步的,所述辐照剂量为8kgy。
27.有益效果:本发明的一种青黛辐照前后主要成分的测定方法,对青黛辐照前后的质量进行研究,对保证产品的质量和临床疗效有着重要意义。
附图说明
28.图1是本发明实施例2中不同检测波长下的青黛hplc指纹图谱色谱图;
29.图2是本发明实施例2中不同流动相系统下的青黛hplc指纹图谱色谱图;
30.图3是本发明实施例2中不同流动相梯度下的青黛hplc指纹图谱色谱图;
31.图4是本发明实施例2中不同流速下的青黛hplc指纹图谱色谱图;
32.图5是本发明实施例2中3批青黛辐照前后样品hplc指纹图谱色谱图;
33.图6是本发明实施例2中青黛hplc对照指纹图谱;
34.图7是本发明实施例4中青黛样品放置6个月整体化学成分稳定性考察hplc指纹图谱;
35.图8是本发明实施例4中青黛样品放置7个月整体化学成分稳定性考察hplc指纹图谱;
36.图9是本发明实施例4中青黛样品放置8个月整体化学成分稳定性考察hplc指纹图谱。
具体实施方式
37.本发明所用仪器包括:agilent 1260高效液相色谱仪(四元泵、自动进样器、柱温箱、dad检测器)(美国安捷伦公司);kq
‑
5200型超声波清洗器(昆山市超声仪器有限公司);hh数显恒温水浴锅(江苏金坛市金城国胜实验仪器厂);at201型十万分之一电子天平(梅特勒
‑
托利多仪器有限公司);精密天平(xp6型,梅特勒
‑
托利多仪器有限公司);fa2104分析电子天平(上海良平仪器仪表有限公司)。
38.本发明所用试剂与药材包括:乙腈(hplc,tedia);甲醇(hplc,tedia);磷酸(色谱纯,南京化学试剂股份有限公司);水为millipore超纯水。
39.本发明所用对照品:靛蓝(中国食品药品检定研究院,批号中国食品药品检定研究院,批号110716
‑
201612,含量以98.7%计);靛玉红(中国食品药品检定研究院,批号110717
‑
201805,含量以99.6%计)。辐照前后青黛样品由江苏七〇七天然制药有限公司提供。辐照剂量为8kgy。样品信息见表1。辐照前缩写为bi(before irradiation),辐照后缩写为ai(after irradiation),附在样品批号后,本发明中所有样品均采用此缩写。
40.表1 青黛样品信息
[0041][0042]
实施例1
[0043]
青黛辐照前后靛蓝和靛玉红含量的测定
[0044]
对照品溶液的制备:取靛蓝或靛玉红对照品2.5mg,精密称定,置250ml容量瓶中,加2%水合氯醛的三氯甲烷溶液(取水合氯醛,置硅胶干燥器中放置24小时,称取2.0g,加三氯甲烷至100ml,放置,出现浑浊,以无水硫酸钠脱水,滤过,即得)约220ml,超声处理(功率250w,频率33khz)1.5小时,放冷,加2%水合氯醛的三氯甲烷溶液至刻度,摇匀,即得(每l ml中含靛蓝或靛玉红10μg);
[0045]
供试品溶液的制备:取青黛样品50mg,精密称定,置250ml容量瓶中,加n,n
‑
二甲基甲酰胺约220ml,超声处理(功率250w,频率33khz)30分钟,放冷,加n,n
‑
二甲基甲酰胺至刻度,摇匀,滤过,取续滤液,即得;
[0046]
色谱条件:色谱柱:agilent zorbax sb
‑
c18(4.6mm
×
250mm,5μm),保护柱:agilent c18 ods(4.6mm
×
12.5mm,5μm),以甲醇
‑
水(70:30)为流动相,检测波长为292nm,柱温,25℃,进样量:10μl;
[0047]
样品分析:取3批次辐照前后样品s1~s3,分别平行制备对照品和供试品溶液各2份,色谱条件下平行测定2次,记录峰面积,采用外标一点法计算样品中靛蓝和靛玉红含量。
[0048]
3批青黛药材辐照前后靛蓝和靛玉红含量结果见表2。
[0049]
表2 青黛辐照前后靛蓝和靛玉红含量
[0050][0051][0052]
表2中,相对含量变化为辐照后与辐照前结果相比较,+表示辐照后含量升高,
‑
表示辐照后含量降低。
[0053][0054]
公式中m
辐照后
为辐照后青黛药材中靛蓝、靛玉红含量,m
辐照前
为未辐照青黛药材中靛蓝、靛玉红含量。
[0055]
2015版《中国药典》中青黛药材的含量测定要求,按干燥品计算,含靛蓝不得少于2.0%,含靛玉红不得少于0.13%,测得的结果显示,各批次辐照前后药材中靛蓝和靛玉红含量均符合药典规定。比较同批次辐照前后青黛样品中靛蓝和靛玉红含量结果,按照公式1计算青黛辐照前后靛蓝和靛玉红相对含量变化百分比,结果见表2,由结果可知,3批样品辐照前后,靛玉红含量无变化,靛蓝相对含量变化在3%~8%之间;进一步对3批样品辐照前后靛蓝含量进行配对样本t检验,可得,p
靛蓝
=0.068>0.05。由上述结果可知,在8kgy的辐照剂量下青黛药材经辐照后靛蓝和靛玉红的含量与辐照前无显著性差异。
[0056]
实施例2
[0057]
青黛辐照前后整体化学成分的测定
[0058]
对照品溶液的制备:分别取靛蓝、靛玉红对照品1.0mg,精密称定,加甲醇制成每
1ml含0.1mg的溶液,即得;
[0059]
供试品溶液的制备:取青黛粉末250mg,精密称定,置具塞锥形瓶中,精密加入甲醇25ml,称定重量,超声提取30min,放冷,再称定重量,称重后甲醇补足失重,摇匀滤过,取续滤液,即得;
[0060]
色谱条件:色谱柱:agilent zorbax sb
‑
c18(4.6mm
×
250mm,5μm),保护柱agilent zorbax sb
‑
c18(4.6mm
×
12.5mm,5μm);流动相:乙腈(b)
‑‑
0.02%磷酸水(ph=3)(a),梯度洗脱:0~5min,20%b;5~35min,20%~55%b;35~45min,55%~66%b;45~60min,66%~95%b;流速为0.80ml
·
min
‑1;检测波长为242nm;柱温30℃;进样量20μl;
[0061]
指纹图谱测定:精密吸取对照品溶液和供试品溶液各20μl,注入高效液相色谱仪,测定,得到各批次样品色谱图和各色谱峰峰面积,比较同批次辐照后样品与未辐照样品之间的差异;
[0062]
指纹图谱的建立:取3批次辐照前后青黛样品s1~s3共计6份样品,测定指纹图谱,将s1设为参照谱,时间宽度为0.10,采用中位数法,生成对照图谱,确定其中14个色谱峰为青黛的指纹图谱共有峰,建立指纹图谱,如图5所示,并以39.99min左右处出峰的靛玉红为参照峰s,取3批次青黛药材辐照前后样品和对照药材指纹图谱计算各色谱峰保留时间和保留峰面积与同一图谱中s峰的保留时间和保留峰面积的比值,得到相对保留时间和相对峰面积,比较特征峰即图谱的相似度。
[0063]
结果表明,各共有峰的相对保留时间rsd值均小于5%,说明共有峰出峰时间较稳定,相对峰面积结果见表3,各共有峰相对峰面积rsd值在5.77%~41.95%之间,说明不同生产批次青黛样品各成分含量存在一定的差异。各图谱与对照图谱的相似度分别为0.994、0.986、0.997、0.999、0.995、0.999,均大于0.95,相似度较高。
[0064]
以3批次辐照前样品建立指纹图谱,每批样品与对照指纹图谱相似度分别为0.988、0.998、1.000;以3批次辐照后样品建立指纹图谱,每批样品与对照指纹图谱相似度分别为0.985、0.999、1.000;3批辐照后样品与辐照前样品对照指纹图谱相似度分别为0.986、0.998、0.999;辐照前样品对照指纹图谱与辐照后样品对照指纹图谱相似度为0.985。
[0065]
3批次辐照前与其相应辐照后样品相似度分别为0.988、0.998、0.996,均大于0.95。
[0066]
由以上结果可知,无论以3批辐照前后样品建立指纹图谱共有模式还是分别以辐照前和辐照后样品建立指纹图谱共有模式,各批样品与对照指纹图谱相似度均大于0.95,辐照前样品与辐照后样品间相似度也均大于0.95,表明辐照前后样品中成分种类基本无变化,辐照对青黛整体化学成分影响较小,不同批次青黛样品整体一致性较好。
[0067]
表3 3批辐照前后青黛药材指纹图谱共有峰相对峰面积
[0068][0069]
实施例3
[0070]
青黛辐照前后样品中靛蓝和靛玉红含量稳定性考察
[0071]
取3批次辐照前后青黛样品s1~s3,放置0、6、7、8个月后,采用实施例1中青黛含量测定方法对辐照前后靛蓝、靛玉红含量(%)进行测定,以此判断辐照对青黛药材中靛蓝、靛玉红含量稳定性的影响,结果分别见表4和表5。
[0072]
表4 青黛辐照前后靛蓝含量(%)稳定性考察
[0073][0074]
表5 青黛辐照前后靛玉红含量(%)稳定性考察
[0075]
[0076]
由结果可见,3批次样品s1、s2、s3辐照后放置6、7、8个月后,靛蓝含量变化rsd值在3.9%~9.5%之间,靛玉红含量变化rsd值在2.1%~3.5%之间;对辐照前后0、6、7、8个月样品中靛蓝、靛玉红含量进行配对样本t检验,结果见表6。由表可知,放置6、7、8个月时,靛蓝含量t检验p值均大于0.05,说明靛蓝含量虽有变化但变化无显著差异;靛玉红含量在6个月和7个月时t检验的p值均大于0.05,8个月时由于含量差值的标准误差为0,无法进行t检验。综上可知,辐照对青黛药材稳定性无显著影响。
[0077]
表6 青黛辐照前后靛蓝含量稳定性t检验结果
[0078][0079][0080]
表6中,
“‑”
是由于差值的标准误差为0,无法进行t检验。
[0081]
实施例4
[0082]
青黛辐照前后样品整体化学成分稳定性考察
[0083]
取3批次辐照前后青黛样品s1~s3,放置0、6、7、8个月后,采用实施例2中青黛指纹图谱分析方法对辐照前后青黛样品进行指纹图谱分析,以此判断辐照对青黛样品整体化学成分稳定性的影响。为便于指纹图谱稳定性分析,其中s1~s6为0个月时3批次辐照前后样品,s7~s12为放置不同月份(6、7、8)后上述3批次辐照前后样品编号,奇数编号代表辐照前样品,偶数编号代表辐照后样品,样品编号详见表7。
[0084]
表7 青黛稳定性考察样品信息
[0085][0086]
青黛样品放置6个月后,样品分析结果见图7,根据《中药色谱指纹图谱相似度评价
系统》(2004a版)对放置0个月与6个月样品谱图进行相似度计算,放置0个月与6个月各批次样品相似度分别为0.974、0.992、0.989,均大于0.95,表明样品放置6个月后整体化学成分无明显变化。
[0087]
青黛样品放置7个月后,样品分析结果见图8,根据《中药色谱指纹图谱相似度评价系统》(2004a版)对放置0个月与7个月样品谱图进行相似度计算,放置0个月与7个月各批次样品相似度分别为0.977、0.989、0.990,均大于0.95,表明样品放置7个月后整体化学成分无明显变化。
[0088]
青黛样品放置8个月后,样品分析结果见图9。根据《中药色谱指纹图谱相似度评价系统》(2004a版)对放置0个月与8个月样品谱图进行相似度计算,放置0个月与8个月各批次样品相似度分别为0.976、0.989、0.989,均大于0.95,表明样品放置8个月后整体化学成分无明显变化。
[0089]
通过以上研究数据表明,青黛通过钴60辐照,既保证了青黛的微生物限度符合散剂入药的要求,又保留了药物的有效成分,确保临床使用疗效。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1