一种二噁英类化合物的检测方法与流程
1.本发明属于环境与食品检测技术领域,尤其涉及一种二噁英类化合物的检测方法。
背景技术:
2.二噁英类化合物一般是指多氯代二苯并二噁英(pcdds)及多氯代二苯并呋喃(pcdfs)的总称(简称pcdd/fs),是一类世界已知的有毒化合物中毒性最强的。通常,二噁英类化合物在环境中较难分解,水中的溶解度较低,生物富集性高。另外,在环境中常常对大气、土壤、河流、湖泊、海洋等造成严重污染,并且它能沿着食物链达到顶层的动物体内,可影响人类生殖和发育、损害免疫系统、干扰激素稳态、具有致癌性,并且在人体内会长期蓄积。
3.现有针对水体中二噁英类化合物的检测方法存在针对“生活饮用水”中二噁英类化合物检测的适宜性和针对性不足,并且检测灵敏度和数据表达的规范性要求不够精确,无法满足对“生活饮用水”中二噁英类化合物的限量标准要求,以及前处理净化方法繁琐,需消耗大量原料溶剂,耗时长的问题。
技术实现要素:
4.本发明实施例的目的在于提供一种二噁英类化合物的检测方法,旨在解决现有的二噁英类化合物的检测方法存在针对“生活饮用水”中二噁英类化合物检测的适宜性和针对性不足,并且检测灵敏度和数据表达的规范性要求不够精确,无法满足对“生活饮用水”中二噁英类化合物的限量标准要求,以及前处理净化方法繁琐,需消耗大量原料溶剂,耗时长的问题。
5.本发明实施例是这样实现的,一种二噁英类化合物的检测方法,包括:
6.s1:在待测饮用水样品中加入提取内标后,采用二氯甲烷进行振摇萃取,并对所得萃取液进行浓缩处理,得浓缩萃取液;
7.s2:将所述浓缩萃取液加入到酸性硅胶层析柱后,采用正己烷洗脱、浓缩,得第一浓缩洗脱液;
8.s3:将所述第一浓缩洗脱液加入到中性氧化铝层析柱后,依次采用正己烷、二氯甲烷洗脱、浓缩,得第二浓缩洗脱液;
9.s4:将所述第二浓缩洗脱液加入到活性炭层析柱后,依次采用正己烷与二氯甲烷的混合溶液、甲苯洗脱、浓缩,得第三浓缩洗脱液;
10.s5:将所述第三浓缩洗脱液用二氯甲烷溶解,经氮气吹扫浓缩后,加入壬烷以及多氯代二苯并二噁英/多氯代二苯并呋喃回收率内标标准溶液,采用高分辨气相色谱/高分辨双聚焦磁式质谱联用仪进行分析检测,确定所述待测饮用水样品中二噁英类化合物的含量。
11.本发明实施例提供的二噁英类化合物的检测方法,采用液
‑
液萃取方法提取饮用
水中的17种pcdd/fs,萃取液经过酸性硅胶柱、氧化铝柱净化以及活性炭柱分离与浓缩定容后,用高分辨气相色谱/高分辨双聚焦磁式质谱联用仪进行分析检测,同位素稀释内标法准确定量生活饮用水中17种pcdd/fs化合物的含量。本发明对生活饮用水中二噁英类化合物检测的针对性强,与现有检测方法比较,其净化处理过程简单、所用溶剂和材料配制种类少、消耗溶剂量少、有毒有害试剂暴露低、耗时短以及灵敏度高,满足对生活饮用水中二噁英类化合物的限量标准要求。
附图说明
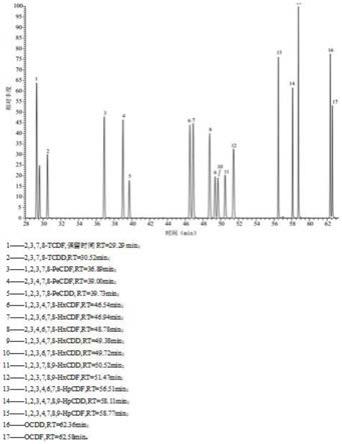
12.图1是本发明实施例提供的17种pcdd/fs目标化合物的总离子流色谱图;
13.图2是本发明实施例提供的2,3,7,8
‑
tcdd分离度检查色谱图。
具体实施方式
14.为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
15.在本发明实施例中,17种pcdd/fs的最低检测质量浓度分别为:2,3,7,8
‑
四氯代二苯并二噁英(2,3,7,8
‑
tcdd)最低检测质量浓度为1.2pg/l;2,3,7,8
‑
四氯代二苯并呋喃(2,3,7,8
‑
tcdf)最低检测质量浓度为1.2pg/l;1,2,3,7,8
‑
五氯代二苯并二噁英(1,2,3,7,8
‑
pecdd)最低检测质量浓度为3pg/l;1,2,3,7,8
‑
五氯代二苯并呋喃(1,2,3,7,8
‑
pecdf)最低检测质量浓度为6pg/l;2,3,4,7,8
‑
五氯代二苯并呋喃(2,3,4,7,8
‑
pecdf)最低检测质量浓度为9pg/l;1,2,3,4,7,8
‑
六氯代二苯并二噁英(1,2,3,4,7,8
‑
hxcdd)最低检测质量浓度为6pg/l;1,2,3,6,7,8
‑
六氯代二苯并二噁英(1,2,3,6,7,8
‑
hxcdd)最低检测质量浓度为9pg/l;1,2,3,7,8,9
‑
六氯代二苯并二噁英(1,2,3,7,8,9
‑
hxcdd)最低检测质量浓度为6pg/l;1,2,3,4,7,8
‑
六氯代二苯并呋喃(1,2,3,4,7,8
‑
hxcdf)最低检测质量浓度为6pg/l;1,2,3,6,7,8
‑
六氯代二苯并呋喃(1,2,3,6,7,8
‑
hxcdf)最低检测质量浓度为12pg/l;1,2,3,7,8,9
‑
六氯代二苯并呋喃(1,2,3,7,8,9
‑
hxcdf)最低检测质量浓度为3pg/l;2,3,4,6,7,8
‑
六氯代二苯并呋喃(2,3,4,6,7,8
‑
hxcdf)最低检测质量浓度为6pg/l;1,2,3,4,6,7,8
‑
七氯代二苯并二噁英(1,2,3,4,6,7,8
‑
hpcdd)最低检测质量浓度为6pg/l;1,2,3,4,6,7,8
‑
七氯代二苯并呋喃(1,2,3,4,6,7,8
‑
hpcdf)最低检测质量浓度为6pg/l;1,2,3,4,7,8,9
‑
七氯代二苯并呋喃(1,2,3,4,7,8,9
‑
hpcdf)最低检测质量浓度为3pg/l;八氯代二苯并二噁英(ocdd)最低检测质量浓度为21pg/l;八氯代二苯并呋喃(ocdf)最低检测质量浓度为15pg/l。适用于生活饮用水中二噁英类化合物的测定。
16.在本发明实施例中,采用液
‑
液萃取方法提取生活饮用水中的17种pcdd/fs,萃取液经过酸性硅胶柱、氧化铝柱净化以及活性炭柱分离与浓缩定容后,用高分辨气相色谱/高分辨双聚焦磁式质谱联用仪(hrgc/hrms)进行分析检测,同位素稀释内标法准确定量生活饮用水中17种pcdd/fs化合物的含量。根据目标化合物的毒性当量因子(tef)与所测得的质量浓度相乘后累加,计算样品中17种pcdd/fs化合物的毒性当量浓度(teq)。17种pcdd/fs化合物和其毒性当量因子见表1。
17.表1
[0018][0019][0020]
在本发明实施例中,检测过程中所用试剂和材料包括:正己烷(c6h
14
);二氯甲烷(ch2cl2);丙酮(c3h6o);甲醇(ch3oh);甲苯(c7h8);壬烷(c9h
20
);浓硫酸(h2so4):优级纯;无水硫酸钠(na2so4):优级纯,在550℃下处理6h,在干燥器中密封保存;玻璃棉:农残级;全氟煤油(pfk)或全氟三丁胺(fc43):用于高分辨双聚焦磁式质谱仪器校正和质量数的校准;超纯水(h2o):经过二级纯化的实验室用水,并使用本发明方法不得检出pcdd/fs。除非另有说明,本发明涉及的水均为经过该处理后的水。以上有机溶剂均为农残级,浓缩10000倍后不得检出二噁英及其类似物。
[0021]
在本发明实施例中,pcdd/fs同位素标记定量内标标准溶液为用壬烷配制的
13
c
12
‑
pcdd/fs溶液,浓度见表2,测试前用50倍丙酮稀释,当天稀释当天使用。
[0022]
表2
[0023]
同位素标记的化合物浓度/(ng/ml)同位素标记的化合物浓度/(ng/ml)
13
c
12
‑
2,3,7,8
‑
tcdd100
13
c
12
‑
1,2,3,6,7,8
‑
hxcdf100
13
c
12
‑
2,3,7,8
‑
tcdf100
13
c
12
‑
1,2,3,7,8,9
‑
hxcdf100
13
c
12
‑
1,2,3,7,8
‑
pecdd100
13
c
12
‑
2,3,4,6,7,8
‑
hxcdf100
13
c
12
‑
1,2,3,7,8
‑
pecdf100
13
c
12
‑
1,2,3,4,6,7,8
‑
hpcdd100
13
c
12
‑
2,3,4,7,8
‑
pecdf100
13
c
12
‑
1,2,3,4,6,7,8
‑
hpcdf100
13
c
12
‑
1,2,3,4,7,8
‑
hxcdd100
13
c
12
‑
1,2,3,4,7,8,9
‑
hpcdf100
13
c
12
‑
1,2,3,6,7,8
‑
hxcdd100
13
c
‑
ocdd200
13
c
12
‑
1,2,3,4,7,8
‑
hxcdf100
‑‑
[0024]
在本发明实施例中,pcdd/fs同位素标记回收率内标标准溶液为用壬烷配制的
13
c
12
‑
1,2,3,4
‑
tcdd和
13
c
12
‑
1,2,3,7,8,9
‑
hxcdd溶液,浓度见表3。
[0025]
表3
[0026]
同位素标记的回收率内标浓度/(ng/ml)
13
c
12
‑
1,2,3,4
‑
tcdd200
±
10
13
c
12
‑
1,2,3,7,8,9
‑
hxcdd200
±
10
[0027]
在本发明实施例中,pcdd/fs精密度和回收率(par)检查标准溶液为用壬烷配制的含有天然17种pcdd/fs化合物的溶液,浓度见表4,用于方法建立时的初始精密度和回收率(ipr)试验及过程精密度和回收率(opr)试验。
[0028]
表4
[0029]
天然化合物浓度/(ng/ml)天然化合物浓度/(ng/ml)2,3,7,8
‑
tcdd401,2,3,6,7,8
‑
hxcdf2002,3,7,8
‑
tcdf401,2,3,7,8,9
‑
hxcdf2001,2,3,7,8
‑
pecdd2002,3,4,6,7,8
‑
hxcdf2001,2,3,7,8
‑
pecdf2001,2,3,4,6,7,8
‑
hpcdd2002,3,4,7,8
‑
pecdf2001,2,3,4,6,7,8
‑
hpcdf2001,2,3,4,7,8
‑
hxcdd2001,2,3,4,7,8,9
‑
hpcdf2001,2,3,6,7,8
‑
hxcdd200ocdd4001,2,3,7,8,9
‑
hxcdd200ocdf4001,2,3,4,7,8
‑
hxcdf200
‑‑
[0030]
在本发明实施例中,pcdd/fs校正标准(cs)溶液为含有天然和同位素标记的pcdd/fs系列校正溶液,浓度见表5。测定校正标准溶液,可以获得天然与同位素标记pcdd/fs的相对响应因子(rrf)或响应因子(rf)。此外,cs3用于已建立rrf/rf的日常校正和校正曲线校验(ver);cs1可用于检查hrgc/hrms应具备的灵敏度。
[0031]
表5
[0032][0033][0034]
在本发明实施例中,时间窗口确定和分离度检查标准溶液用于时间窗口确定和2,3,7,8
‑
tcdd分离度的检查,浓度见表6。用壬烷配制,为含有同位素标记pcdd/fs(定量内标和回收率内标)的溶液,用于方法的校正和确证,并可以用于db
‑
5ms毛细管柱(或等效柱)中四氯至八氯取代化合物出峰顺序确定,同时用于检查在规定的色谱柱中2,3,7,8
‑
tcdd和2,3,7,8
‑
tcdf的分离度。
[0035]
表6
[0036][0037][0038]
在本发明实施例中,样品净化所使用的吸附剂包括:活性硅胶:0.063mm~0.200mm(粒径),或相当等级的硅胶。在使用前,分别用两倍体积的甲醇、二氯甲烷清洗后,于180℃烘烤至少1h,在干燥器中冷却,保存在带螺帽密封的玻璃瓶中,2周内用完;酸性硅胶(44%,质量分数):称取56g活性硅胶置于250ml具塞磨口旋转烧瓶中,在玻璃棒搅拌下用滴管逐滴加入44g浓硫酸,再将烧瓶加塞后置于摇床上进行振荡,直至硅胶呈均匀流动状态,在干燥器中贮存,2周内用完;中性氧化铝:0.063mm~0.200mm(粒径),或相当等级的中性氧化铝。在使用之前,于550℃马弗炉内烘烤至少2h,在干燥器中贮存;混合活性炭(18%,质量分数):称取9.0g carbopak c(或其它相当的类型)和41.0g celite 545(或其它相当的类型)充分混合,于130℃烘箱中活化4h~6h,在干燥器中贮存。
[0039]
本发明实施例提供的二噁英类化合物的检测方法,具体包括以下步骤:
[0040]
s1:在待测饮用水样品中加入提取内标后,采用二氯甲烷进行振摇萃取,并对所得萃取液进行浓缩处理,得浓缩萃取液。
[0041]
在本发明实施例中,所述步骤s1,具体包括:
[0042]
s11:在待测饮用水样品中加入丙酮稀释好的
13
c
12
标记定量内标溶液。
[0043]
s12:在待测饮用水样品中加入二氯甲烷,经振摇萃取、静置分层后,转移出有机相,经过无水硫酸钠脱水后合并萃取液,并将所得萃取液进行浓缩处理,得浓缩萃取液。
[0044]
在本发明实施例中,所述步骤s1中,所述待测饮用水样品与二氯甲烷的体积比为(2~10):1。
[0045]
s2:将所述浓缩萃取液加入到酸性硅胶层析柱后,采用正己烷洗脱、浓缩,得第一浓缩洗脱液。
[0046]
在本发明实施例中,步骤s2中,所述酸性硅胶层析柱为从下到上按照0.05g~0.2g无水硫酸钠、0.5g~5g酸性硅胶以及0.05g~0.2g无水硫酸钠的顺序干法装填形成;所述酸性硅胶层析柱内各组分在装填之后采用正己烷预冲洗。其中,经本发明前期实验发现,若酸性硅胶量不足,影响样品的净化效果,增加仪器检测分析的难度;若酸性硅胶量过大,会增加试剂的消耗量和延长净化时间。
[0047]
s3:将所述第一浓缩洗脱液加入到中性氧化铝层析柱后,依次采用正己烷、二氯甲烷洗脱、浓缩,得第二浓缩洗脱液。
[0048]
在本发明实施例中,步骤s3中,所述中性氧化铝层析柱为从下到上按照0.05g~0.2g无水硫酸钠、1g~4g中性氧化铝以及0.05g~0.2g无水硫酸钠的顺序干法装填形成;所述中性氧化铝层析柱内各组分在装填之后采用正己烷预冲洗。其中,经本发明前期实验发现,若中性氧化铝的含量不足,影响样品的净化效果,增加仪器检测分析的难度;若中性氧化铝的含量过大,会增加试剂的消耗量和延长净化时间。
[0049]
s4:将所述第二浓缩洗脱液加入到活性炭层析柱后,依次采用正己烷与二氯甲烷的混合溶液、甲苯洗脱、浓缩,得第三浓缩洗脱液。
[0050]
在本发明实施例中,步骤s4中,所述活性炭层析柱为从下到上按照0.05g~0.2g无水硫酸钠、0.1g~2g 18%炭粉以及0.05g~0.2g无水硫酸钠的顺序干法装填形成;所述中性氧化铝层析柱内各组分在装填之后先采用正己烷预冲洗,再依次加入二氯甲烷、甲苯、正己烷与二氯甲烷的混合溶液以及正己烷进行淋洗。其中,经本发明前期实验发现,若18%炭粉的添加量不足,会减低样品的回收率;若18%炭粉的添加量过量,会增加试剂的消耗量和大大延长净化时间。另外,甲苯洗脱过程中,使用40ml~60ml甲苯对富集在炭粉中的目标物进行洗脱,若洗脱用甲苯体积不足,目标物ocdd的回收率会会偏低。
[0051]
在本发明实施例中,步骤s4中,所述正己烷与二氯甲烷的混合溶液中正己烷与二氯甲烷二者的体积比为1:(0.5~2)。
[0052]
s5:将所述第三浓缩洗脱液用二氯甲烷溶解,经氮气吹扫浓缩后,加入壬烷以及多氯代二苯并二噁英/多氯代二苯并呋喃回收率内标标准溶液,采用高分辨气相色谱/高分辨双聚焦磁式质谱联用仪进行分析检测,确定所述待测饮用水样品中二噁英类化合物的含量。
[0053]
在本发明实施例中,步骤s5中,采用高分辨气相色谱/高分辨双聚焦磁式质谱联用仪检测时,色谱条件为:毛细管柱:db
‑
5ms柱,60m
×
0.25mm
×
0.25μm或等效色谱柱;进样口温度:260℃~300℃;进样方式:不分流;进样体积:1~3μl;传输线温度:260℃~290℃;升温程序:100℃~140℃保持0.5min~2min,以30℃/min~60℃/min升至210℃~230℃,保持10min~25min;再以0.5℃/min~2.5℃/min升至240℃~254℃,以0.25℃/min~0.6℃/min升至255℃~270℃;最后以30℃~50℃/min升至280℃~320℃,以280℃~320℃保持5~
15min;载气:氦气,0.5ml/min~1.5ml/min。
[0054]
在本发明实施例中,步骤s5中,采用高分辨气相色谱/高分辨双聚焦磁式质谱联用仪检测时,质谱条件为:电离模式:电子轰击;扫描模式:选择性离子监测;分辨率≥10000。
[0055]
在本发明实施例中,步骤s5中,所述多氯代二苯并二噁英/多氯代二苯并呋喃回收率内标标准溶液为用壬烷配制的
13
c
12
‑
1,2,3,4
‑
四氯代二苯并二噁英和
13
c
12
‑
1,2,3,7,8,9
‑
六氯代二苯并二噁英溶液。
[0056]
下面结合具体实施例详细说明本发明检测方法,但并不应认为是限制本发明的定义。
[0057]
s1:样品前处理
[0058]
准确量取1l的水样,然后将水样全部转移至2l的分液漏斗中,并加入500μl丙酮稀释好的
13
c
12
标记定量内标溶液。在样品中加入200ml二氯甲烷,振摇萃取20min(振摇期间注意放气)。静置分层后,转移出有机相。重复上述操作,继续连续萃取水样2次,每次使用200ml二氯甲烷。经过无水硫酸钠脱水后合并萃取液。用旋转蒸发仪浓缩萃取液至1ml~2ml,以进行后续净化。
[0059]
s2:酸性硅胶柱净化
[0060]
层析柱的填充:取内径为5mm的玻璃层析柱,底部填充少量玻璃棉,依次装入0.1g无水硫酸钠,1g酸性硅胶,0.1g无水硫酸钠。干法装柱,轻敲层析柱,使吸附剂分布均匀。用10ml正己烷预淋洗层析柱,弃去淋洗液。向层析柱加入浓缩萃取液后,使用15ml正己烷分3次清洗玻璃瓶并加载在层析柱上,然后使用20ml正己烷进行洗脱,并收集洗脱液。将收集的洗脱液浓缩至1ml~2ml。
[0061]
s3:中性氧化铝净化
[0062]
层析柱的填充:取内径为5mm的玻璃层析柱,底部填充少量玻璃棉,依次装入0.1g无水硫酸钠,1.5g中性氧化铝,0.1g无水硫酸钠。干法装柱,轻敲层析柱,使吸附剂分布均匀。使用10ml正己烷预淋洗层析柱,弃去淋洗液。向层析柱加入浓缩洗脱液,使用15ml正己烷分3次清洗玻璃瓶并加载到层析柱上,然后使用10ml正己烷淋洗层析柱,并弃去淋洗液。使用10ml二氯甲烷进行洗脱,并收集洗脱液。将收集的洗脱液浓缩至1ml~2ml。
[0063]
s4:活性炭柱净化
[0064]
层析柱的填充:取内径为5mm的玻璃层析柱,底部填充少量玻璃棉,依次装入0.1g无水硫酸钠,0.15g 18%炭粉,0.1g无水硫酸钠。干法装柱,轻敲层析柱,使吸附剂分布均匀。先使用10ml正己烷预淋洗层析柱。然后依次加入1ml二氯甲烷、10ml甲苯、1ml正己烷:二氯甲烷(1:1,体积比)和2ml正己烷进行淋洗,弃去淋洗液。向层析柱加入浓缩洗脱液,使用15ml正己烷分3次清洗玻璃瓶并加载到玻璃层析柱上,再加入3ml正己烷:二氯甲烷(1:1,体积比)进行洗脱,弃去洗脱液。然后加入40ml甲苯进行洗脱,并收集洗脱液。将收集的洗脱液浓缩至近干。
[0065]
s5:微量浓缩
[0066]
将收集瓶内样品浓缩液用二氯甲烷溶解分次转移到装有100μl内插管的进样小瓶,并使用10ml二氯甲烷清洗浓缩瓶至少5次,在高纯氮气(99.999%)吹扫下微量浓缩至近干,加入15μl壬烷,再加入5μlpcdd/fs回收率内标标准溶液,混匀后待进样hrgc/hrms分析检测。
[0067]
其中,pcdd/fs分析条件:
[0068]
色谱条件:
[0069]
毛细管柱:db
‑
5ms柱(5%苯基
‑
甲基聚硅氧烷),60m
×
0.25mm
×
0.25μm或等效色谱柱。进样口温度:280℃。进样方式:不分流。进样体积:2μl。传输线温度:280℃。升温程序:120℃保持1min,以50℃/min升至220℃,保持15min;再以1.5℃/min升至250℃,以0.59℃/min升至260℃;最后以33℃/min升至310℃,以310℃保持8min。载气:高纯氦气(99.999%),1.0ml/min(恒流)。
[0070]
质谱条件:
[0071]
电离模式:电子轰击(ei);扫描模式:选择性离子监测(sim)。
[0072]
质量校正:采用质量校准物质(全氟煤油,pfk;或全氟三丁胺,fc43)的质量数锁定进行质量偏移校正。pfk在m/z=304.9824或fc43在m/z=313.9839或者其他接近的参考气离子碎片,调节进入hrms中参考气的量使锁定质量数的峰信号强度不得超过检测器量程的10%,然后进行分辨率调谐,调整质谱以满足最小所需的10000分辨率(10%峰谷分离)。分辨率应≥10000,精确的m/z与表7中的理论m/z之间的偏差应小于百万分之五。
[0073]
分辨率:在分辨率≥10000的条件下,监测表7规定的各个化合物两个精确质量数的离子,得到其选择离子流图。
[0074]
在给定的gc条件下,进样1μl或2μl cs1校正溶液,考察离子丰度比、最小水平、信噪比及绝对保留时间:
[0075]
1)测定各目标化合物峰面积,计算表7中规定的精确质量数离子的丰度比,并符合表8理论值中的质量控制范围要求;否则需要调谐,重新测定,使其符合规定。各窗口需要进行连续监测,确保在gc运行中能够监测全部的pcdd/fs化合物单体。各窗口监测的精确质量数离子详见表7。
[0076]
2)各化合物的保留时间与其定量参考化合物单体间的相对保留时间的偏差应符合表9规定的范围。
[0077]
3)hrgc/hrms应满足表10仪器检出限要求,进样标准系列的cs1时pcdd/fs单体化合物和标记化合物的信噪比(s/n)≥10。
[0078]
4)
13
c
12
‑
1,2,3,4
‑
tcdd在db
‑
5ms柱上的绝对保留时间应大于25.0min。
[0079]
表7
[0080]
[0081]
[0082][0083]
表8
[0084][0085]
表9
[0086]
[0087][0088]
表10
[0089]
化合物名称仪器检出限/(pg/μl)方法检出限/(pg/l)最低检测质量浓度/(pg/l)2,3,7,8
‑
tcdd0.040.41.22,3,7,8
‑
tcdf0.030.41.21,2,3,7,8
‑
pecdd0.1131,2,3,7,8
‑
pecdf0.1262,3,4,7,8
‑
pecdf0.09391,2,3,4,7,8
‑
hxcdd0.2261,2,3,6,7,8
‑
hxcdd0.1391,2,3,7,8,9
‑
hxcdd0.07261,2,3,4,7,8
‑
hxcdf0.1261,2,3,6,7,8
‑
hxcdf0.14121,2,3,7,8,9
‑
hxcdf0.1132,3,4,6,7,8
‑
hxcdf0.1261,2,3,4,6,7,8
‑
hpcdd0.09261,2,3,4,6,7,8
‑
hpcdf0.1261,2,3,4,7,8,9
‑
hpcdf0.113ocdd0.3721ocdf0.2515
[0090]
异构体的确认:
[0091]
a)采用时间窗口确定和分离度检查标准溶液,进行异构体确认。所划分的四氯、五氯、六氯、七氯和八氯取代的pcdd/fs窗口的总离子流色谱图见图1。
[0092]
b)gc分辨率检查:计算距离2,3,7,8
‑
tcdd最近异构体的gc色谱峰谷高度,不应超
过25%。如果化合物间峰谷高度超过25%,应调整分析条件并重新测试或更换gc柱,进行校正。按照式(1)计算峰谷高比,2,3,7,8
‑
tcdd分离度检查色谱图见图2。
[0093][0094]
式中:hv——峰谷高比;x——2,3,7,8
‑
tcdd与1,2,3,7/1,2,3,8
‑
tcdd的峰谷高度,单位为毫米(mm);y——2,3,7,8
‑
tcdd的峰高,单位为毫米(mm)。
[0095]
同位素稀释法校正:
[0096]
1)同位素稀释内标法适用于15种在样品萃取前加入
13
c
12
标记同位素定量内标的pcdd/fs单体。
[0097]
2)根据表7中第一个和第二个精确质量数离子的响应峰面积,按式(2)计算各化合物相对于其标记化合物的rrf。
[0098][0099]
式中:rrf——相对响应因子;a1
n
和a2
n
——校正标准中pcdd/fs的第一个和第二个监测离子的峰面积;a1
l
和a2
l
——校正标准中
13
c
12
标记定量内标化合物的第一个和第二个监测离子的峰面积;c
n
——校正标准中天然目标化合物的浓度,单位为ng/ml;c
l
——校正标准中
13
c
12
标记定量内标化合物的浓度,单位为ng/ml。
[0100]
3)在给定的条件下测定5个校正标准溶液(cs1~cs5)中各目标化合物的rrf。
[0101]
4)如果每种化合物在5点校正曲线的rrf的相对标准偏差(rsd)≤20%,则对该化合物可以采用平均响应因子进行计算,否则需要重新校正。
[0102]
内标法校正:
[0103]
1)适用于检测1,2,3,7,8,9
‑
hxcdd、ocdf以及
13
c
12
标记同位素化合物。
[0104]
2)响应因子(rf):
[0105]
a)1,2,3,7,8,9
‑
hxcdd与其定量内标之间的响应因子。根据表7中第一个和第二个精确质量数离子的响应峰面积,按照式(3)计算1,2,3,7,8,9
‑
hxcdd的rf
h
。
[0106][0107]
式中:rf
h
——1,2,3,7,8,9
‑
hxcdd的响应因子;a1
h
和a2
h
——校正标准中1,2,3,7,8,9
‑
hxcdd的第一个和第二个质量数离子的峰面积;a1
h1
和a2
h1
——校正标准中
13
c
12
‑
1,2,3,4,7,8
‑
hxcdd的第一个和第二个质量数离子的峰面积;a1
h2
和a2
h2
——校正标准中
13
c
12
‑
1,2,3,6,7,8
‑
hxcdd的第一个和第二个质量数离子的峰面积;c
h
——校正标准中1,2,3,7,8,9
‑
hxcdd的浓度,单位为ng/ml;c
h1
和c
h2
——校正标准中
13
c
12
‑
1,2,3,4,7,8
‑
hxcdd和
13
c
12
‑
1,2,3,6,7,8
‑
hxcdd的浓度,单位为ng/ml。
[0108]
b)ocdf与其定量内标(
13
c
12
‑
ocdd)之间的响应因子。根据表7中第一个和第二个精确质量数离子的响应峰面积,按照式(4)计算ocdf的rf
o
。
[0109][0110]
式中:rf
o
——ocdf的响应因子;a1
n
和a2
n
——校正标准中ocdf的第一个和第二个监测离子的峰面积;a1
l
和a2
l
——校正标准中
13
c
12
‑
ocdd的第一个和第二个监测离子的峰面
积;c
n
——校正标准中ocdf的浓度,单位为ng/ml;c
l
——校正标准中
13
c
12
‑
ocdd的浓度,单位为ng/ml。
[0111]
c)
13
c
12
标记定量内标化合物的响应因子。根据表7中第一个和第二个精确质量数离子的响应峰面积,按照式(5)计算各定量内标标记化合物相对于回收率内标标记化合物的响应因子(rf)。
[0112][0113]
式中:rf——
13
c
12
标记定量内标化合物的响应因子;a1
l
和a2
l
——校正标准
13
c
12
标记定量化合物的第一个和第二个监测离子的峰面积;a1
rs
和a2
rs
——校正标准回收率内标化合物的第一个和第二个监测离子的峰面积;c
l
——校正标准中定量内标化合物的浓度,单位为ng/ml;c
rs
——校正标准中回收率内标化合物的浓度,单位为ng/ml。
[0114]
3)在给定的条件下测定5个校正标准溶液(cs1~cs5)中各目标化合物的rrf,采用内标法计算各标准溶液中目标化合物的rf。
[0115]
4)如果每种化合物在5点校正曲线的rf值的相对标准偏差(rsd)≤35%,则对该化合物可以采用平均响应因子进行计算;否则需要重新校正。
[0116]
联合校正:
[0117]
进样含天然未标记pcdd/fs化合物、标记化合物及内标的校正溶液,由同位素稀释和内标方法获取校正曲线。如果满足要求,则采用平均响应因子进行计算;如果不能满足要求,则需要重新校正。
[0118]
仪器校正和运行检查:
[0119]
1)性能检查:
[0120]
在分析过程中,每隔12h校验一次hrgc/hrms仪器性能。以cs3校正标准溶液进样,检查分析系统的各项性能指标。只有在满足下述条件的情况下,才能进行样品检测。
[0121]
a)化合物pcdd/fs的氯同位素分子离子,其离子丰度比应在理论离子丰度比范围内,离子丰度比详见表8。
[0122]
b)标准溶液所检测到的每一个pcdd/fs的质量色谱峰的s/n≥10。
[0123]
c)中点校正标准的非标记pcdd/fs的rrf值的偏移(rsd)应≤20%,
13
c1标记pcdd/fs同系物的rf值偏移应≤35%。
[0124]
d)中点校正标准的进样内标的绝对保留时间应该在校正标准系列的平均保留时间的
±
5s内。
[0125]
2)ms分辨率
[0126]
分析前,确认ms静态分辨率≥10000,并每隔12h检查一次。一旦分辨率达不到要求,则需要重新进行仪器调谐。
[0127]
3)校正标准的验证
[0128]
a)检查各目标化合物离子的丰度比是否满足表8的要求,如果不符合,则需要调谐质谱仪重新校正。
[0129]
b)校正标准中pcdd/fs及其同位素标记化合物色谱峰的信噪比(s/n)≥10,否则需要调谐质谱仪重新校正。
[0130]
c)校正标准中相对响应因子之间的相对标准偏差≤20%,响应因子之间的相对标
准偏差≤35%。如果不符合要求,则需要调谐质谱仪重新校正。
[0131]
4)实验室本底污染检查
[0132]
在开展试验之前进行实验室全程空白实验,对实验系统本底污染情况进行检查。采用超纯水作为空白基质进行与实际样品相同的实验流程,检查实验室pcdd/fs空白本底水平,以确定分析系统未受到污染,其中四氯~八氯取代pcdd/fs不得检出,不得高于方法检出限。
[0133]
5)分析起始和过程中的精密度及回收率试验
[0134]
a)在方法建立时或重要物质和高分辨质谱仪有重大改变时,应首先进行分析精密度及回收率(opr)试验。在超纯水中加入pcdd/fs的精密度和回收率检查标准溶液使其在最终提取液中pcdd/fs的浓度达到表11中的测试浓度范围。
[0135]
b)采用同位素稀释法计算天然未标记pcdd/fs化合物含量;使用内标法计算1,2,3,7,8,9
‑
hxcdd和ocdf单体的含量,同时计算15种
13
c
12
标记同位素内标化合物的回收率,回收率范围符合表12规定范围,才可进行实际样品分析。
[0136]
表11
[0137][0138][0139]
表12
[0140][0141]
初始精密度和回收率检查:
[0142]
在实验室开始使用本方法进行实际样品分析前需要对实验室的分析能力进行检查。采用本方法的实验步骤进行萃取、净化和浓缩,分析4个添加
13
c
12
同位素标记定量内标和ipr的超纯水样品,使其最终提取液(20μl)中pcdd/fs的浓度达到表11中的测试浓度。计算4个ipr样品中pcdd/fs的平均浓度(x)和标准偏差s,并计算
13
c
12
标记定量内标的回收率。当所测定pcdd/fs化合物的平均值与标准偏差与其相应的
13
c
12
标记同位素内标回收率分别符合表11和表12规定的范围时,表明实验室具备了分析能力,可以开展实际样品的分析。
[0143]
试验数据处理:
[0144]
1)定性分析:
[0145]
根据精确质量测定的两个质量碎片离子的理论丰度比和保留时间进行定性。所监测的两个精确的m/z信号,信号在2s内达到最大值,同时监测的两个分子离子精确质量的峰面积比值应符合表8的规定,或在校正标准cs3中相应两个质量数离子的峰面积比值的
±
10%范围内。各目标化合物的相对保留时间应符合表9规定。
[0146]
样品提取物所检测到的每一个pcdd/fs的gc峰的s/n≥2.5,校正标准溶液各化合物的s/n≥10。
[0147]
检测pcdd/fs时,尚需检查分离度,确保2,3,7,8
‑
tcdd与其相邻的最接近的同分异构体峰谷的高度小于全峰高的25%。
[0148]
当上述定性指标未达到要求时,应进一步检查色谱质谱的性能,或者重新净化样品,除去干扰物质后重新进样分析。
[0149]
2)定量分析:
[0150]
a)浓度计算:
[0151]
同位素稀释内标法定量:
[0152]
适用于除1,2,3,7,8,9
‑
hxcdd和ocdf外的其它pcdd/fs的定量。
[0153]
按照公式(6)计算样品中目标化合物的浓度:
[0154][0155]
式中:c——样品中pcdd/fs的浓度,单位为pg/l;rrf
av
——平均相对响应因子;a1
n
和a2
n
——pcdd/fs的第一个和第二个监测离子的峰面积;a1
l
和a2
l
——
13
c
12
定量内标的第
一个和第二个监测离子的峰面积;c
l
——样品提取前加入的
13
c
12
标记的定量内标量,单位为pg;v——样品取样体积,单位为l。
[0156]
1,2,3,7,8,9
‑
hxcdd的定量:
[0157]
由于
13
c
12
‑
1,2,3,7,8,9
‑
hxcdd作为回收率内标,因此1,2,3,7,8,9
‑
hxcdd的定量不能采用严格的同位素稀释法,而采用
13
c
12
‑
1,2,3,4,7,8
‑
hxcdd及
13
c
12
‑
1,2,3,6,7,8
‑
hxcdd平均响应因子定量计算。
[0158]
按照公式(7)计算样品中1,2,3,7,8,9
‑
hxcdd的浓度:
[0159][0160]
式中:c
h
——样品中1,2,3,7,8,9
‑
hxcdd的浓度,单位为pg/l;a1
h
和a2
h
——1,2,3,7,8,9
‑
hxcdd的第一个和第二个质量数离子的峰面积;a1
h1
和a2
h1
——
13
c
12
‑
1,2,3,4,7,8
‑
hxcdd的第一个和第二个质量数离子的峰面积;a1
h2
和a2
h2
——
13
c
12
‑
1,2,3,6,7,8
‑
hxcdd的第一个和第二个质量数离子的峰面积;c
h1
和c
h2
——样品提取前加入的
13
c
12
‑
1,2,3,4,7,8
‑
hxcdd和
13
c
12
‑
1,2,3,6,7,8
‑
hxcdd的量,单位为pg;rf
hav
——1,2,3,7,8,9
‑
hxcdd的平均响应因子;v——样品取样体积,单位为l。
[0161]
ocdf的定量:
[0162]
由于潜在的干扰问题,没有向样品中加入ocdf的同位素标记物,因此ocdf由
13
c
12
同位素标记的ocdd定量。
[0163]
按照公式(8)计算样品中ocdf的浓度:
[0164][0165]
式中:c
o
——样品中ocdf的浓度,单位为pg/l;a1
n
和a2
n
——ocdf的第一个和第二个监测离子的峰面积;a1
l
和a2
l
——
13
c
12
‑
ocdd的第一个和第二个监测离子的峰面积;c
l
——样品提取前加入
13
c
12
‑
ocdd的量,单位为pg;rf
oav
——ocdf的平均响应因子;v——样品取样体积,单位为l。
[0166]
回收率计算:
[0167]
a)样品提取液中
13
c
12
标记定量内标的计算,按照公式(9)计算:
[0168][0169]
式中:m
ex
——样品
13
c
12
标记定量内标的测定结果,单位为pg;a1
l
和a2
l
——
13
c
12
标记定量内标化合物的第一个和第二个监测离子的峰面积;a1
rs
和a2
rs
——回收率内标化合物的第一个和第二个监测离子的峰面积;c
rs
——进样前加入的回收率内标的量,单位为pg;rf
av
——平均响应因子。
[0170]
b)依据上述定量结果,按照公式(10)计算
13
c
12
标记定量内标的回收率:
[0171][0172]
式中:x——样品回收率,%;m
ex
——样品
13
c
12
标记定量内标的测定结果,单位为pg;m
l
——样品提取加入的
13
c
12
标记定量内标的量,单位为pg。
[0173]
毒性当量计算:
[0174]
按照世界卫生组织(who)规定的pcdd/fs的毒性当量因子(tef)和公式(11~14)计算样品中pcdd/fs的毒性当量(teq),其中各化合物tef值见表1。
[0175]
teq
i
=tef
i
×
c
i
ꢀꢀꢀ
(11)
[0176]
teq
pcdds
=∑tef
ipcdds
×
c
ipcdds
ꢀꢀꢀ
(12)
[0177]
teq
pcdfs
=∑tef
ipcdfs
×
c
ipcdfs
ꢀꢀꢀ
(13)
[0178]
teq
pcdd/fs
=teq
pcdds
+teq
pcdfs
ꢀꢀꢀ
(14)
[0179]
式中:teq
i
——样品中pcdd/fs的毒性当量浓度,单位为pg teq/l;tef
i
——pcdd/fs的毒性当量因子;c
i
——样品中pcdd/fs的质量浓度,单位为pg/l;teq
pcdds
——样品中pcdd的毒性当量浓度,单位为pg teq/l;teq
pcdfs
——样品中pcdfs的毒性当量浓度,单位为pg teq/l;teq
pcdd/fs
——样品中pcdd/fs的毒性当量浓度,单位为pg teq/l。
[0180]
检出限计算:
[0181]
仪器检出限:
[0182]
采用系列标准溶液中最低质量浓度的标准溶液(cs1)进样质谱仪连续测定5次以上,计算5次测定值的标准偏差s,取标准偏差的3倍(3s),修约为1位或2位有效数字作为仪器检出限。本发明的仪器检出限见表10。
[0183]
方法检出限:
[0184]
使用超纯水中加标方法来计算方法检出限(mdl)。按照本方法,在纯水中添加标准物质(par),添加绝对量为国家限量标准的1/30,即在1l超纯水中,添加1pg 2,3,7,8
‑
tcdd;然后进行与样品处理相同的提取、净化、浓缩和仪器分析方法,进行定性和准确定量。重复上述测定5次,计算测定值的标准偏差,取标准偏差的3倍,结果修约为1位或2位有效数字作为方法检出限。取方法检出限的3倍,获得最低检测质量浓度,本标准的方法检出限和最低检测质量浓度见表10。
[0185]
结果报告:
[0186]
1)样品中的pcdd/fs需要报告质量浓度包括7种pcdds化合物、10种pcdfs化合物以及17种单体化合物合计质量浓度、检出限和毒性当量值(teq),包括teq
pcdds
、teq
pcdfs
和teq
pcdd/fs
。
[0187]
2)报告检出限参考国家标准《数值修约规则和极限数值的表示和判定》(gb/t 8170
‑
2008)中修约为1位或2位有效数字。质量浓度和teq结果大于100时,只保留整数部分;结果介于1和100之间时,结果保留三位有效数字;结果低于1时,结果保留2位有效数字。浓度和teq结果的小数位数应不多于检出限位数。
[0188]
3)对于样品的实测质量浓度,在检出限或以上的结果,以实际检测结果报告浓度。低于检出限的结果可以报告“未检出”(n.d.)或按照管理机构的要求报告。另外,根据需要可以报告总pcdd/fs、总pcdds和总pcdfs的质量浓度,对于低于检出限的质量浓度,如无特别指明,按照0来计算pcdd/fs、pcdds和pcdfs的合计质量浓度。
[0189]
4)对于毒性当量,为实测质量浓度与相应毒性当量因子(tef)的乘积。对于低于检出限的化合物单体质量浓度,如无特别指明,使用检出限计算相应单体毒性当量。
[0190]
回收率、精密度和准确度测定:
[0191]
在生活饮用水中(本底值为未检出,取样量为1l)加入pcdd/fs精密度和回收率检查标准溶液(par)进行低、中、高三个浓度水平的实验,使2,3,7,8
‑
tcdd加入par的绝对量分
别为1pg,30pg和45pg,每个浓度梯度平行分析5次。17种pcdd/fs回收率情况详见表13,回收率均符合表12要求。准确度以加标回收率进行评价;精密度以加标回收率的相对标准偏差(rsd)进行评价,如表13所示。
[0192]
表13
[0193][0194][0195]
注:n.d.未检出。
[0196]
应用实例:
[0197]
在本发明的实际应用中发现,采用200ml二氯甲烷萃取水样获得的样品回收率最佳。具体操作如下:在生活饮用水中(本底值为未检出,取样量为1l)加入中浓度的pcdd/fs精密度和回收率检查标准溶液(par),使2,3,7,8
‑
tcdd加入par的绝对量分别为30pg,分别在等量的不同生活饮用水样品中加入50ml、100ml、150ml、200ml和250ml二氯甲烷,振摇萃取20min(振摇期间注意放气)。静置分层后,转移出有机相。重复上述操作,继续连续萃取水样2次,每次使用相应等量的二氯甲烷,分别对每个二氯甲烷萃取体积平行分析5次。17种
pcdd/fs回收率情况详见表14。
[0198]
表14
[0199]
[0200][0201][0202]
在生活饮用水中(取样量为1l)加入pcdd/fs精密度和回收率检查标准溶液(par)
进行低、中、高三个浓度水平的实验,使2,3,7,8
‑
tcdd加入par的绝对量分别为1pg,30pg和45pg,每个浓度梯度平行分析6次,重复7次实验。实验结果均符合表12的范围,详见表15。
[0203]
表15
[0204][0205][0206]
综上,本发明实施例提供的二噁英类化合物的检测方法,采用液
‑
液萃取方法提取饮用水中的17种pcdd/fs,萃取液经过酸性硅胶柱、氧化铝柱净化以及活性炭柱分离与浓缩定容后,用高分辨气相色谱/高分辨双聚焦磁式质谱联用仪进行分析检测,同位素稀释内标法准确定量生活饮用水中17种pcdd/fs化合物的含量。本发明对生活饮用水中二噁英类化合物检测的针对性强,相较比现有检测方法,其净化处理过程简单、所用溶剂和材料配制种
类少、消耗溶剂量少、有毒有害试剂暴露低、耗时短以及灵敏度高,满足对生活饮用水中二噁英类化合物的限量标准要求。
[0207]
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1