本发明涉及环保
技术领域:
,更具体地说是一种gc-ms测定大气干湿沉降物中多环芳烃的方法。
背景技术:
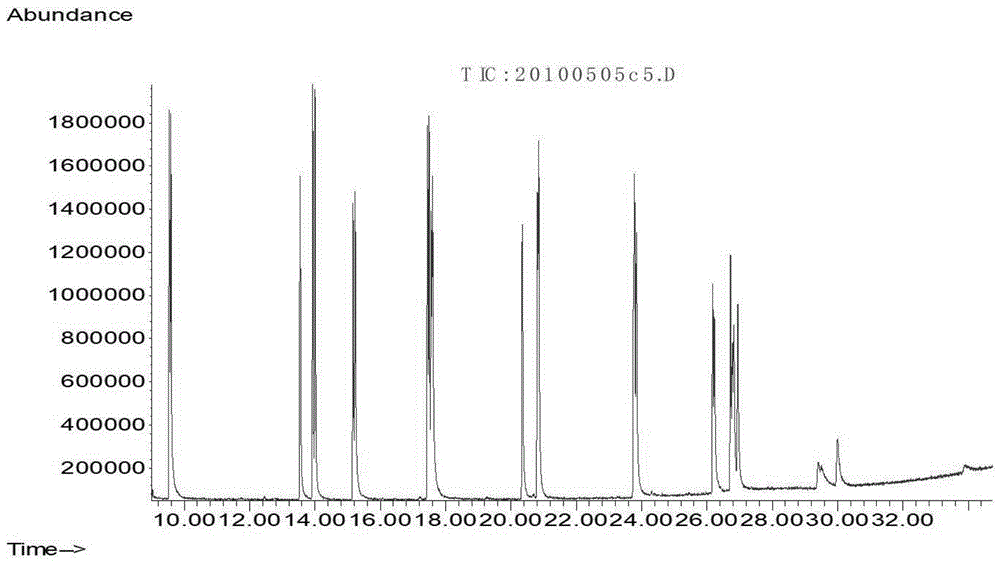
:大气干湿沉降是土壤污染的途径之一,大气干湿沉降物中的多环芳烃,是反映大气尘粒污染的主要指标之一。多环芳烃的产生既有天然源,也有人为源。陆地和水生植物、微生物的生物合成,森林、草原的天然火灾以及火山活动所形成的多环芳烃构成了多环芳烃的天然本底值。由于细菌活动和植物腐烂所形成的土壤多环芳烃本底值为100~1000μg/kg。地下水中多环芳烃的本底值为0.001~0.01μg/l。淡水湖泊中的本底值为0.01~0.25μg/l。人为源多环芳烃的污染源很多,它可以在我们生活的每一个角落发现,任何有有机物加工废弃,燃烧或使用的地方都有可能产生多环芳烃,例如炼油厂、炼焦厂、橡胶厂和火电厂等任何一家排放烟尘的工厂,各种交通车辆排放的尾气中、煤气及其他取暖设施甚至居民的炊烟中等。多环芳烃有三方面特性:第一,持久性。多环芳烃通过各种环境介质(大气、水、生物体等)能够长距离迁移并长期存在于环境中,进而对人类健康和环境带来严重的危害。由于多环芳烃的水溶性极小,它们在土壤中的降解和生物可利用性受到严重限制,由于其具有较高的辛醇-水分配系数,易于分配到环境疏水性有机物中,因此在生物体脂类中易于富集浓缩,有较高的生物富集因子。第二,“三致”作用。多环芳烃类化合物具有强烈的致突变作用、致癌作用和致畸作用,简称“三致”作用。其对动植物的生长都有明显的影响,多环芳烃落在植物叶片上,使其变色、萎缩、卷曲、直至脱落,影响植物的正常生长和结果。多环芳烃对动物的影响也较严重,随食物摄入人体内的苯并[α]芘大部分可被人体吸收,经过消化道吸收后,经过血液很快遍布人体,人体吸收的苯并芘一部分与蛋白质结合,另一部分则参与代谢分解,与蛋白质结合的苯并芘可与亲电子的细胞受体结合,使制细胞生长的酶发生变异,使细胞失去控制生长的能力而发生癌变。第三,生物蓄积性。多环芳烃进入环境后以通过环境蓄积、生物蓄积、生物转化或化学反应等方式损害健康和环境,多环芳烃并不是直接致癌物,它在体内经过酶的作用后生成终致癌物。经致癌物与dna或rna等结合后产生不可修复的损害而导致癌症。目前,国外测定大气中多环芳烃的方法有很多,iso12884:2000《环境空气吸附剂-滤膜采集气相和颗粒物中的多环芳烃气相色谱-质谱法》、usepa标准:《空气中有毒有机化合物测定方法汇编》第二版pa/625/r-96/010b)中的汇编方法to-13a《气相色谱-质谱联机法测定环境空气中的多环芳烃》、方法8270e《气相色谱法/质谱分析法测试半挥发性有机化合物》(2018年6月6日修订)测定环境空气和废气、水和废水以及土壤中的多环芳烃,使用毛细管柱分离,气相色谱质谱(gc-ms)检测。国内也有一些相关的检测方法,如《hj834-2017土壤和沉积物半挥发性有机物的测定气相色谱-质谱法》《hj805-2016土壤和沉积物多环芳烃的测定气相色谱-质谱法》《hj646-2013环境空气和废气气相和颗粒物中多环芳烃的测定气相色谱-质谱法》中均有多环芳烃的测定方法,前处理外色谱条件等相关内容可参考其进行。但是,关于大气干湿沉降物中多环芳烃的检测方法未见报道。技术实现要素:目前,开展大气干湿沉降物中多环芳烃的检测方法未见报道。为满足检测需要,本方法参照《hj805-2016土壤和沉积物多环芳烃的测定气相色谱-质谱法》《hj646-2013环境空气和废气气相和颗粒物中多环芳烃的测定气相色谱-质谱法》,首次提出建立gc-ms测定大气干湿沉降物中16种多环芳烃的检测方法。为实现上述目的,本发明提供如下技术方案:一种gc-ms测定大气干湿沉降物中多环芳烃的方法,包括以下步骤:将大气沉降物样品,经索氏提取或加速溶剂萃取后,萃取液干燥、浓缩、硅胶净化后,样品浓缩液进行气相色谱质谱(gc-ms)的全扫描检测,然后通过目标组分的质谱图和保留时间与计算机谱库中的质谱图和保留时间作对照进行定性,每个定性出来组分的浓度取决于其定量离子与内标物定量离子的质谱响应之比;最后每个样品中加入已知浓度的内标化合物,用内标法定量分析。提取的方法为:将适量大气沉降物样品和10.0g无水硫酸钠混合,加入适量替代物,放于提取套管中;用100ml正己烷:丙酮(v:v=1:1)提取液提取12h;待提取液冷却后,将其转移至500ml的分液漏斗中,加入适量的2%的硫酸钠溶液洗去丙酮。脱水,旋蒸浓缩至约1ml,待净化。净化的方法为:a.固相萃取柱的活化在固相萃取柱上端填入约0.5cm的无水硫酸钠,依次用15ml二氯甲烷和10ml正己烷淋洗小柱,当无水硫酸钠即将露出时关闭活塞;该步骤操作过程中不能让无水硫酸钠暴露在空气中,如果该情况发生需要重新条件化小柱;b.样品的转移和净化将浓缩后的样品无损地转移至固相萃取柱中,用另外的1ml正己烷完成全部化合物的转移,转移完成后保持5min后打开活塞,弃去该部分流出液,当无水硫酸钠即将露出时关闭活塞;c.样品的洗脱用洗脱液(1.6)20ml分几次洗脱固相萃取柱,分次用10ml氮吹管盛接该流出液;d.浓缩定容在40℃下氮吹浓缩3.1.3.3流出液至略少于500.00μl,用微量注射器定容500.00μl,加入内标10.0μl,摇匀后上机测定。色谱条件为:进样口温度:280℃;不分流进样,进样量2μl;进样口压力9.95psi,吹扫流量11.0ml/min,吹扫时间2min;色谱柱流量1.1ml/min,流速39cm/sec;升温程序:70℃保持4min,以10℃/min的速率升温至300℃,保持2min,以5℃/min的速率升温至340℃保持直至所有组分流出;接口温度:285℃。质谱条件为:调谐方式:dfttp;扫描方式:全扫描,扫描范围50-450amu;离子源温度:230℃;四极杆温度150℃;电离能量:70ev。标准曲线的绘制a.校准系列的制备:吸取适量标准储备液和内标储备液用正己烷定容至10ml,配置多环芳烃化合物的浓度为1000、400、200、100、50、20ng/ml的校准点,每个校准点的内标浓度为400ng/ml;b.初始校准曲线:按设定的数据采集方法进行数据采集,以内标/目标化合物的浓度为横坐标,内标/目标离子的响应为纵坐标,内标法绘制校准曲线,曲线的相关系数大于0.995,各化合物响应因子的相对标准偏差(rsd)不大于30%,否则重新绘制校准曲线;c.绘制标准样品的总离子流图质谱的校准方法为:开机稳定2小时后按tfapp方式调谐质谱仪,按设定方法注入十氟三苯基磷2μl;按设定方法分析至少5个校准点绘制工作曲线;样品分析:按校准曲线相同的仪器条件下分析处理好的样品;空白实验:在分析每批样品时应作空白试验,检查分析过程中是否有污染。数据处理和结果计算:提取液中化合物浓度的计算:提取离子并积分峰面积,按公式q=c*v/n计算化合物含量,式中:q-样品的含量,ng/g;c-校准曲线计算出的提取液浓度,ng/ml;v-测试提取液的体积,ml;n-样品的取样量,g。还包括重复分析:重复分析样品数量不低于5%,分析项目合格率应大于94%,重复测定不满足要求时,应扩大抽查比例直至满足要求。为满足我国生态环境保护需要,建立gc-ms测定于大气干湿沉降物中16种多环芳烃的检测方法。本方法为首次提出,且符合我国国土资源、生态环境相关标准的要求,不与其他标准存在冲突。本方法的实施,将对中华人民共和国《环境保护法》、农业地质调查、城市地质调查等国家法律法规中有关多环芳烃类化合物污染检测提供可靠的技术段。根据本方法的技术特点和实际应用情况分析总结,本方法有以下优点:(1)检测流程短,精密度和准确度良好,可用于大批量检测工作。(2)当取样量为1.0g时,方法检出限为:0.30~0.60ng/g,满足实际样品的分析要求。(3)为大气干湿沉降物中多环芳烃分析提供了切实可行的方法,填补了大气干湿沉降物中多环芳烃分析的技术空白。附图说明图1为本发明标准样品的总离子流图。具体实施方式下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。实施例一一种gc-ms测定大气干湿沉降物中多环芳烃的方法,包括以下步骤:将大气沉降物样品,经索氏提取(或加速溶剂萃取)后,萃取液干燥、浓缩、硅胶净化后,样品浓缩液进行气相色谱质谱(gc-ms)的全扫描检测。通过目标组分的质谱图和保留时间与计算机谱库中的质谱图和保留时间作对照进行定性,每个定性出来组分的浓度取决于其定量离子与内标物定量离子的质谱响应之比。每个样品中加入已知浓度的内标化合物,用内标法定量分析。16种多环芳烃的名称见表1。表118种pahs的名称(含两种替代物)实施例二一、试剂和材料本方法所使用的试剂除另有注明外,均应符合国家标准分析纯化学试剂。1.1二氯甲烷(ch2cl2):农残级或相当级别。1.2正己烷(c6h14):农残级或相当级别。1.3丙酮(ch3coch3):农残级或相当级别。1.4标准溶液1.4.1多环芳烃标准储备液:浓度为10mg/l,包括表1中16种化合物,用正己烷稀释至2mg/l,4℃以下保存。1.4.2内标物标准储备液:浓度为1000mg/l,包括苝-d12、屈-d12、二氢苊-d12、萘-d8、菲-d10,正己烷稀释至20mg/l,4℃以下保存。1.4.3替代物标准储备液:浓度为500mg/l,包括三联苯-d14、4-溴-2-氟联苯,正己烷稀释至20mg/l,4℃以下保存。1.5调谐液:十氟三苯基磷(dftpp),正己烷介质浓度为2mg/l,4℃以下保存。1.6洗脱液:二氯甲烷与正己烷按体积比1:1混合。1.7固相萃取柱:2.0g10ml硅胶柱,采用其它规格时需要验证。1.8无水硫酸钠:(na2so4)400℃烘烤4h,玻璃瓶中密封保存。二、仪器和设备2.1气相色谱-质谱联用仪(gc-msd)2.2色谱柱:db-5ms,30m,液膜厚0.25μm,内径0.25mm,其它性能相近的色谱柱也可采用。2.3浓缩装置:自动氮吹浓缩仪,或性能相当设备。2.4可控温旋转蒸发仪。2.5可控温恒温水浴装置。2.6其它实验室一般设备。三、分析步骤3.1样品的处理3.1.1提取将适量大气沉降物样品和10.0g无水硫酸钠混合,加入适量替代物,放于提取套管中。用100ml正己烷:丙酮(v:v=1:1)提取液提取12h。待提取液冷却后,将其转移至500ml的分液漏斗中,加入适量的2%的硫酸钠溶液洗去丙酮。脱水,旋蒸浓缩至约1ml,待净化。3.1.3净化3.1.3.1固相萃取柱的活化:在固相萃取柱上端填入约0.5cm的无水硫酸钠,依次用15ml二氯甲烷和10ml正己烷淋洗小柱,当无水硫酸钠即将露出时关闭活塞。该步骤操作过程中不能让无水硫酸钠暴露在空气中,如果该情况发生需要重新条件化小柱。3.1.3.2样品的转移和净化将浓缩后的样品无损地转移至固相萃取柱中,用另外的1ml正己烷完成全部化合物的转移,转移完成后保持5min后打开活塞,弃去该部分流出液,当无水硫酸钠即将露出时关闭活塞。3.1.3.3样品的洗脱用洗脱液(1.6)20ml分几次洗脱固相萃取柱,分次用10ml氮吹管盛接该流出液。3.1.3.4浓缩定容在40℃下氮吹浓缩3.1.3.3流出液至略少于500.00μl,用微量注射器定容500.00μl,加入内标10.0μl,摇匀后上机测定。3.2色谱条件3.2.1进样口温度:280℃;不分流进样,进样量2μl;进样口压力9.95psi,吹扫流量11.0ml/min,吹扫时间2min。3.2.2色谱柱流量1.1ml/min,流速39cm/sec。3.2.3升温程序:70℃保持4min,以10℃/min的速率升温至300℃,保持2min,以5℃/min的速率升温至340℃保持直至所有组分流出。3.2.4接口温度:285℃。3.3质谱条件3.3.1调谐方式:dfttp3.3.2扫描方式:全扫描,扫描范围50-450amu。3.3.3离子源温度:230℃;四极杆温度150℃。3.3.4电离能量:70ev。3.4标准曲线的绘制3.4.1校准系列的制备吸取适量标准储备液和内标储备液用正己烷定容至10ml,配置多环芳烃化合物的浓度为1000、400、200、100、50、20ng/ml的校准点,每个校准点的内标浓度为400ng/ml。3.4.2初始校准曲线按设定的数据采集方法进行数据采集,以内标/目标化合物的浓度为横坐标,内标/目标离子的响应为纵坐标,内标法绘制校准曲线,曲线的相关系数大于0.995,各化合物响应因子的相对标准偏差(rsd)不大于30%,否则重新绘制校准曲线。优选的,使用多环芳烃内标见表2。化合物定量、限定离子见表3。表2多环芳烃使用的内标物表3化合物定量、限定离子3.4.3如图1所示的标准样品的总离子流图出峰顺序:1)d8-na-9.473)d10-ac-13.8812)d10-phe-17.4016)d12-chr-23.7521)d12-pyl-26.922)na-9.514)acl-13.505)ac-13.966)fl-15.187)bghip-29.968)ip-29.339)dbaha-29.4110)bbfa-26.5511)4-br-2f-15.6913)phe-17.4614)an-17.5615)fa-20.3117)baa-23.7218)chr-23.8119)py-20.8222)bap-26.8020)ter-d14-21.3623)bkfa-26.20。3.5样品的测定3.5.1质谱的校准开机稳定2小时后按tfapp方式调谐质谱仪,按设定方法注入十氟三苯基磷2μl,其离子丰度应满足表4要求。表4dftpp关键离子和离子丰度指标质量数离子丰度指标质量数离子丰度指标51198质量数的30%~60%199198质量数的5%~9%68小于69质量数的2%275198质量数的10%~30%70小于69质量数的2%365大于198质量数的1%127198质量数的40%~60%441出现,但小于443质量数的丰度197小于198质量数的1%442大于198质量数的40%198基峰,相对丰度为100%443442质量数的17%~23%3.5.2按设定方法分析至少5个校准点绘制工作曲线(3.4.2)。3.5.3样品分析:按校准曲线相同的仪器条件下分析处理好的样品(3.1.3.4)。3.6空白实验在分析每批样品时应作空白试验,检查分析过程中是否有污染。四、数据处理和结果计算提取液中化合物浓度的计算:按表3提取离子并积分峰面积,按下式计算化合物含量。q=c*v/n式中:q-样品的含量,ng/g。c-校准曲线计算出的提取液浓度,ng/ml。v-测试提取液的体积,ml。n-样品的取样量,g。五、质量控制和质量保证5.1空白:所有空白测试结果应低于方法检出限5.2试剂空白:每批次试剂至少分析一个空白试验5.3空白试验:每批次样品至少分析一个空白试验5.4加标回收率控制范围:60-130%。5.5连续校准相对误差不超过±20%。5.6dftpp验证频次:15工作日一次。5.7重复分析重复分析样品数量不低于5%,分析项目合格率应大于94%,重复测定不满足要求时,应扩大抽查比例直至满足要求。最后应说明的几点是:首先,在本申请的描述中,需要说明的是,除非另有规定和限定,术语“安装”、“相连”、“连接”应做广义理解,可以是机械连接或电连接,也可以是两个元件内部的连通,可以是直接相连,“上”、“下”、“左”、“右”等仅用于表示相对位置关系,当被描述对象的绝对位置改变,则相对位置关系可能发生改变;以上所述仅为本发明的优选实施例而已,并不用于限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。当前第1页12