液质联用结合超滤法测定美罗培南或亚胺培南的血浆蛋白结合率的制作方法
1.本发明涉及药物分析领域,具体地说,涉及液质联用结合超滤法测定美罗培南或亚胺培南的血浆蛋白结合率的方法。
背景技术:
2.美罗培南(mem)和亚胺培南(imp)为碳青霉烯类抗生素,它们的化学结构不稳定。现有检测美罗培南游离浓度的方法中,由于没有解决药物的不稳定问题,导致测定结果不准确。
技术实现要素:
3.本发明的目的是提供液质联用结合超滤法测定美罗培南或亚胺培南的血浆蛋白结合率。
4.为了实现本发明目的,本发明提供一种液质联用结合超滤法测定美罗培南或亚胺培南的血浆蛋白结合率的方法,包括:
5.1)取空白血浆,加入mops缓冲液,再加入不同浓度的药物溶液,配制成不同浓度的含药血浆样本;再向其中加入内标溶液,涡旋混匀后加入乙腈(乙腈用以沉淀蛋白),涡旋混匀后离心,取上清液加入乙腈混匀,用于hplc
‑
ms/ms检测;以美罗培南或亚胺培南峰面积与内标峰面积之比为纵坐标,美罗培南或亚胺培南的浓度为横坐标,进行回归分析,建立针对血浆中药物总浓度的标准曲线
①
;
6.2)取空白血浆,加入mops缓冲液,将体系转移至超滤管中,于15
‑
30℃6000~10000g离心20
‑
30min,收集空白超滤液;向空白超滤液中加入不同浓度的药物溶液,配制成不同浓度的含药超滤液样本;再向其中加入内标溶液,涡旋混匀后加入乙腈(乙腈用以沉淀蛋白),涡旋混匀后离心,取上清液加入乙腈混匀,用于hplc
‑
ms/ms检测;以美罗培南或亚胺培南峰面积与内标峰面积之比为纵坐标,美罗培南或亚胺培南的浓度为横坐标,进行回归分析,建立针对血浆中游离药物浓度的标准曲线
②
;
7.3)将步骤1)中的空白血浆替换成待测血浆,将待测血浆分成a、b两等份,血浆a用于测定血浆中药物总浓度,血浆b用于测定血浆中游离药物浓度;
8.4)取血浆a,加入mops缓冲液混合,加入内标溶液,涡旋混匀后加入乙腈,涡旋混匀后离心,取上清液加入乙腈混匀,进行hplc
‑
ms/ms检测,测定待测血浆中美罗培南或亚胺培南的峰面积,代入上述标准曲线
①
,得到待测血浆的药物总浓度;
9.5)取血浆b,加入mops缓冲液混合,将体系转移到超滤管中,于15
‑
30℃6000~10000g离心20
‑
30min,收集超滤液;向超滤液中加入内标溶液,涡旋混匀后加入乙腈,涡旋混匀后离心,取上清液加入乙腈混匀,进行hplc
‑
ms/ms检测,测定待测血浆的超滤液中美罗培南或亚胺培南的峰面积,代入上述标准曲线
②
,得到待测血浆的游离药物浓度;按照式i)计算出待测血浆中美罗培南或亚胺培南的血浆蛋白结合率:
10.血浆蛋白结合率=(药物总浓度
‑
游离药物浓度)/药物总浓度
×
100%
ꢀꢀ
式i)
11.其中,步骤1)和2)中所述的空白血浆是指来自同一个体、不含美罗培南或亚胺培南的血浆;
12.所述mops缓冲液的配制方法如下:将3
‑
(n
‑
吗啉)丙磺酸半钠盐2g,溶于10ml水中。
13.所述内标溶液为mem
‑
d6溶液。
14.前述的方法,步骤1)和2)中以mops液为试剂,分别配制浓度为10、20、40、100、200、400、1000μg
·
ml
‑1的美罗培南或亚胺培南标准曲线工作液。
15.所述内标溶液是用mops液配制的mem
‑
d6浓度为50μg
·
ml
‑1的内标溶液。
16.所述mops液的配制方法如下:将3
‑
(n
‑
吗啉)丙磺酸半钠盐0.2g,溶于10ml水中。
17.标准曲线
①
的绘制方法包括:向190μl空白血浆中加入20μl的mops缓冲液,再加入10μl的美罗培南或亚胺培南标准曲线工作液,然后向体系中加入10μl内标溶液,涡旋混匀后加入600μl乙腈,涡旋2min混匀;4℃15000rpm离心5min。取上清液50μl加入80%乙腈200μl混匀,用于hplc
‑
ms/ms检测。
18.标准曲线
②
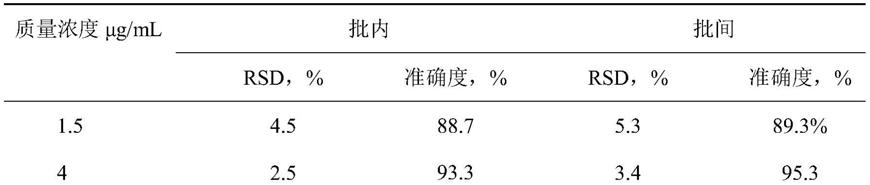
的绘制方法包括:向190μl空白血浆中加入20μl的mops缓冲液,将体系转移至超滤管中,于15~30℃,6000~10000g离心20~30min,收集空白超滤液;取105μl空白超滤液,加入5μl的美罗培南或亚胺培南标准曲线工作液,再加入5μl内标溶液,涡旋混匀后加入300μl乙腈,涡旋2min混匀,取上清液50μl加入80%乙腈200μl混匀,用于hplc
‑
ms/ms检测。
19.步骤4)包括:向200μl待测血浆中加入20μl的mops缓冲液,再加入10μl内标溶液,涡旋混匀后加入600μl乙腈,涡旋2min混匀;4℃15000rpm离心5min。取上清液50μl加入80%乙腈200μl混匀,进行hplc
‑
ms/ms检测。
20.步骤5)包括:向200μl待测血浆中加入20μl的mops缓冲液,将体系转移到超滤管中,于15~30℃6000~10000g离心20~30min,收集超滤液;取110μl超滤液,加入5μl内标溶液,涡旋混匀后加入300μl乙腈,涡旋2min混匀,取上清液50μl加入80%乙腈200μl混匀,进行hplc
‑
ms/ms检测。
21.优选地,色谱检测条件如下:
22.色谱柱:hilic silica,2.1mm
×
50mm,3μm;柱温:40℃;流动相a:含0.1%甲酸的乙腈和含0.1%甲酸的浓度为5mm的甲酸铵水溶液。
23.梯度洗脱程序:0
‑
0.5min,85%a;0.5
‑
2.5min,85%
‑
60%a;2.5
‑
3.5min,60%a;3.5
‑
4.0min,60%
‑
85%a;4.0
‑
6.5min,85%a;流速:0.30ml/min。进样量:0.5μl。
24.优选地,质谱检测条件如下:
25.离子源:电喷雾离子源(esi),正离子扫描方式,离子源温度为550℃,喷雾电压为5500v;多反应监测模式(mrm),监测离子对:美罗培南:384.1
→
141.3,亚胺培南:300
→
126.1,is(mem
‑
d6):390.1
→
147.2;三者碎裂电压分别为22v、15v和24v。
26.前述的方法,当药物为美罗培南时,标准曲线
①
的方程为:y=0.329x
‑
0.00385,r=0.9981;美罗培南的线性检测范围为0.5
‑
50μg/ml,检测下限为0.5μg/ml;
27.标准曲线
②
的方程为:y=0.317x+0.0208,r=0.9982;游离美罗培南的线性检测范围为0.5
‑
50μg/ml。
28.前述的方法,当药物为亚胺培南时,标准曲线
①
的方程为:y=0.227x
‑
0.0124,r=
0.9945;亚胺培南的线性检测范围为0.5
‑
50μg/ml,检测下限为0.5μg/ml;
29.标准曲线
②
的方程为:y=0.287x+0.00316,r=0.9982;游离亚胺培南的线性检测范围为0.5
‑
50μg/ml。
30.优选地,所述超滤管为microcon ym
‑
10centrifugal filter device。
31.借由上述技术方案,本发明至少具有下列优点及有益效果:
32.(一)由于美罗培南、亚胺培南自身的化学结构的不稳定性,本发明在处理样本时加入了mops稳定剂,解决了药物稳定性差的问题。通过引入稳定剂,使蛋白结合更加充分,进而测定结果更加准确。
33.(二)采用本方法测定亚胺培南及美罗培南的游离浓度,设备表面的非特异性吸附不会影响测定准确度,不受稀释效应和体积迁移的影响。由于离心设备可同时实现多样本离心,故可实现批量快速测定。
34.(三)本发明可定量测定不同浓度(低、中、高)的蛋白结合率的差异,应用该方法可比较仿制药在蛋白结合方面的差异,进而评价其一致性。
35.(四)本发明将液相色谱串联质谱检测技术与超滤法相结合,超滤法的优点在于可快速、高通量测定药物的蛋白结合率,尤其适用于不稳定的药物分子。
36.(五)本发明采用蛋白沉淀法处理血浆及超滤液样本,操作简单易行,可实现快速,批量的样本处理。
附图说明
37.图1为本发明较佳实施例中空白血浆色谱图。
38.图2为本发明较佳实施例中空白血浆加内标色谱图。
39.图3为本发明较佳实施例中空白血浆+亚胺培南/美罗培南定量下限浓度的色谱图。
40.图4为本发明较佳实施例中使用symmetry c18柱(2.1
×
50mm,3.5μm)分离的亚胺培南/美罗培南色谱图。
41.图5为本发明较佳实施例中使用atlantis t3柱(2.1
×
50mm,3μm)分离的亚胺培南/美罗培南色谱图。
42.图6为本发明较佳实施例中使用hilic silica柱(2.1mm
×
50mm,3μm),不采用梯度洗脱的亚胺培南/美罗培南色谱图。
43.图7为本发明较佳实施例中使用hilic silica柱(2.1mm
×
50mm,3μm),采用梯度洗脱的亚胺培南/美罗培南色谱图。
具体实施方式
44.以下实施例用于说明本发明,但不用来限制本发明的范围。若未特别指明,实施例中所用的技术手段为本领域技术人员所熟知的常规手段,所用原料均为市售商品。实施例1液质联用结合超滤法测定美罗培南或亚胺培南的血浆蛋白结合率
45.1材料
46.1.1仪器
47.高效液相色谱仪(dgu lc
‑
30ad,日本岛津公司);四极杆/线性离子阱质谱仪
(absciex 4000
+
质谱检测仪,美国ab sciex公司),配有电喷雾离子源(esi);ms3digital涡旋混合器(德国ika公司);bt25s电子天平(德国sartorius公司);3
‑
18k离心机(美国sigma公司);cascada
‑
i纯水机(美国pall公司),hwt
‑
10c(恒奥科技)恒温震荡水浴锅。
48.1.2药品及试剂
49.美罗培南对照品(meropenem,mem),批号:130506
‑
201403,纯度:87.0%,购自中国食品药品检定研究院;亚胺培南对照品(imipenem,imp),批号:fy32693m6762,纯度:98.2%,购自南通飞宇生物科技有限公司;meropenem
‑
d6:批号:m225637,纯度:>90%,购自toronto research chemicals(toronto,canada)。3
‑
(n
‑
吗啉)丙磺酸半钠盐,批号:rqc723,购自上海毕得医药科技有限公司;色谱纯的乙腈购自fisher chemical,hplc级别的甲酸购自fisher scientific,甲酸铵购自sigma公司。超滤管为microcon ym
‑
10centrifugal filter device,购自bedford,usa。
50.2方法
51.2.1溶液配制
52.2.1.1缓冲液(mops)的配制
53.精密称取3
‑
(n
‑
吗啉)丙磺酸半钠盐0.2g,溶于10ml水中,得到2%的mops液,作为用于配制系列标准曲线的工作液;精密称取3
‑
(n
‑
吗啉)丙磺酸半钠盐2g,溶于10ml水中,得到20%的mops缓冲液,作为稳定剂在样本处理过程中添加。
54.2.1.2标准储备液及工作液的配制
55.精密称取mem及imp对照品适量,用2%mops液配制mem及imp质量浓度为2.0mg
·
ml
‑1的标准曲线储备液及2.0mg
·
ml
‑1的质控储备液,再用2%mops液分别稀释配制成浓度为10、20、40、100、200、400、1000μg
·
ml
‑1的标准曲线工作液和浓度为30、80、800μg
·
ml
‑1的质控工作液,定量下限工作液浓度为10μg
·
ml
‑1。精密称取mem
‑
d6对照品适量,用2%mops液配制成质量浓度为1000μg
·
ml
‑1的内标储备液,以上述溶剂稀释成50μg
·
ml
‑1的内标溶液。
56.2.2标准曲线的绘制
57.向190μl空白血浆中加入20μl的20%mops缓冲液,再加入10μl的标准曲线工作液,然后向体系中加入10μl内标溶液,涡旋混匀后加入600μl乙腈,涡旋2min混匀;4℃15000rpm离心5min。取上清液50μl加入80%乙腈200μl混匀,用于hplc
‑
ms/ms检测。以美罗培南或亚胺培南峰面积与内标峰面积之比为纵坐标,美罗培南或亚胺培南的浓度为横坐标,进行回归分析,建立针对血浆中药物总浓度的标准曲线。
58.向190μl空白血浆中加入20μl的20%mops缓冲液,将体系转移到超滤管中,于20℃8000g离心30min,收集空白超滤液;取105μl空白超滤液,加入5μl的美罗培南或亚胺培南标准曲线工作液,再加入5μl内标溶液,涡旋混匀后加入300μl乙腈,涡旋2min混匀,取上清液50μl加入80%乙腈200μl混匀,用于hplc
‑
ms/ms检测。以美罗培南或亚胺培南峰面积与内标峰面积之比为纵坐标,美罗培南或亚胺培南的浓度为横坐标,进行回归分析,建立针对血浆中游离药物浓度的标准曲线。
59.2.3样本前处理
60.2.3.1血浆中药物总浓度测定的样本处理
61.200μl含药血浆样本中加入20μl的20%mops缓冲液。再向体系中加入10μl内标溶液(50μg
·
ml
‑1),涡旋混匀后加入600μl乙腈,涡旋2min混匀;4℃15000rpm离心5min。取上清
液50μl加入80%乙腈200μl混匀,于进样瓶中,进样体积0.5μl。
62.2.3.2超滤样本的处理
63.200μl含药血浆样本中加入20μl的20%mops缓冲液。将体系转移到超滤管中,于20℃8000g离心30min,收集超滤液;取110μl超滤液,加入内标溶液5μl(50μg
·
ml
‑1),涡旋混匀后加入300μl乙腈,涡旋2min混匀,再取上述混合液50μl加入80%乙腈200μl混匀,于进样瓶中,进样体积0.5μl。
64.2.4色谱与质谱条件
65.色谱柱:hilic silica(2.1mm
×
50mm,3μm);柱温:40℃;流动相:乙腈(含0.1%甲酸,a)和5mm甲酸铵水溶液(含0.1%甲酸,b),梯度洗脱程序:0~0.5min,85%a;0.5~2.5min,85%~60%a;2.5~3.5min,60%a;3.5~4.0min,60%~85%a;4.0~6.5min,85%a。进样量:0.5μl;流速:0.30ml/min。
66.离子源:电喷雾离子源(esi),正离子扫描方式,离子源温度为550℃,喷雾电压为5500v;多反应监测模式(mrm),监测离子对:mem:384.1
→
141.3,imp:300
→
126.1,is(mer
‑
d6):390.1
→
147.2;三者碎裂电压分别为22v、15v和24v。
67.3结果
68.3.1专属性验证
69.6个不同来源的空白血浆,分别取190μl,进行如下分组处理:不加内标,加入内标溶液,配制成定量下限样本后加入内标溶液。按“2.3”步骤操作处理后测定,典型色谱图见图1~图3。mem、imp和内标的保留时间分别为2.96、2.85和2.97min。由结果可知,空白血浆对于mem、imp和内标无干扰,内标对于待测物也无干扰。
70.3.2标准曲线与线性检测范围
71.取空白血浆190μl,加入10μl相应浓度的对照品工作液,制备成质量浓度分别为0.5、1、2、5、10、20、50μg/ml的标准曲线样品,按“2.3”步骤处理后测定。以mem或imp与内标峰面积之比为纵坐标(y)、mem或imp的质量浓度为横坐标(x)进行回归分析,权重为1/x2。得美罗培南的回归方程为y=0.329x
‑
0.00385(r=0.9981),亚胺培南的回归方程为:y=0.227x
‑
0.0124(r=0.9945)mem和imp的线性范围均为0.5~50μg/ml,定量下限为0.5μg/ml。
72.超滤液对应的标准曲线方程分别为:mem:y=0.317x+0.0208(r=0.9982);imp:y=0.287x+0.00316(r=0.9982)。游离mem和imp测定的线性范围均为0.5~50μg/ml。
73.3.3精密度与准确度
74.取定量下限工作液及质控工作液分别加入190μl空白血浆,每个浓度平行配制5份,按“2.3”步骤处理后,进样测定。空白血浆同法处理,采用标准加入法考察批内精密度,并以测得量/加入量
×
100%计算批内准确度。同法在3d内配制并测定3批次样品,考察批间精密度和准确度。精密度与准确度结果见表1和表2。
75.表1 mem精密度及准确度结果(n=5)
[0076][0077][0078]
表2 imp精密度及准确度结果(n=5)
[0079][0080]
超滤液精密度与准确度结果见表3和表4。
[0081]
表3超滤液中mem精密度及准确度结果(n=5)
[0082][0083]
表4超滤液中imp精密度及准确度结果(n=5)
[0084][0085]
3.4超滤膜的回收率
[0086]
分别取400μl低、中、高(1.5,4,40μg
·
ml
‑1)3种不同质量浓度的mem及imp水溶液样品置于超滤管中,于4℃,8000g离心20min,取不同浓度mem及imp超滤液110μl,按“2.3”步骤操作处理样品,进样测定超滤液中mem及imp的浓度(ρ
post
)。以及超滤前溶液中药物浓度(ρ
pre
)。
[0087]
回收率=ρ
post
/ρ
pre
×
100%
[0088]
计算出mem及imp的相对回收率,结果见表5和表6。超滤膜的回收率均>96%,表明
mem及imp与超滤膜均无特异性吸附。
[0089]
表5 mem的相对回收率
[0090][0091][0092]
表6 imp的相对回收率
[0093][0094]
3.5孵育稳定性
[0095]
分别配制成含有1.5、4、40μg
·
ml
‑1mem或imp的低、中、高浓度血浆样品加入10%(体积百分数)mops液(浓度为20%的mops液)。将上述不同质量浓度的血浆样品分别置于37℃恒温水浴锅中,振荡频率为100rpm,温育0.5,1,1.5,2h,按“2.3”步骤操作处理样品,测定mem及imp浓度(表7)。结果发现,在上述条件下mem温育2h,血浆样本中美罗培南的含量的保持稳定,imp温育1h,血浆样本中亚胺培南的含量保持稳定。
[0096]
表7
[0097][0098]
3.6超滤透析实验方法
[0099]
取80μl mem(或imp)标准溶液,加入1520μl空白血浆中,再加入160μl的20%mops液,涡旋混匀,制成含有低、中、高(1.5,4,40μg
·
ml
‑1)不同质量浓度的mem(或imp)血浆样品。将上述样品置于37℃恒温水浴锅中,振荡频率为100rpm,温育120min(imp温育60min),各浓度平行3个样本。按“2.3”步骤操作处理样品,按“2.4”色谱条件进样测定,超滤液(cf)与超滤管血浆(cp)中美罗培南(亚胺培南)的浓度,依据以下公式计算药物与血浆蛋白的结合率:
[0100]
结合率=(cp
‑
cf)/cp
×
100%
[0101]
计算出mrm及imp的血浆蛋白结合率。
[0102]
分别测定6批空白血浆与药物孵育后的蛋白结合情况,结果见表8和表9:
[0103]
表8 mem血浆蛋白结合率
[0104][0105]
表9 imp血浆蛋白结合率
[0106][0107]
实施例2离心力及离心时间的优化
[0108]
在超滤管中分别加入200μl及400μl的血浆样本,每体积双样本(编号200
‑
1,200
‑
2及400
‑
1,400
‑
2分别在6000g、8000g及10000g的离心力下,20℃离心20min或30min,考察获得超滤液的体积。结果见表10:
[0109]
表10不同离心条件下超滤液的体积
[0110][0111][0112]
按照不破坏超滤膜,且超滤液与超滤前血浆总体积的比值在0.3~0.6的标准,优选8000g作为离心力,超滤时间在20~30min范围内。
[0113]
实施例3色谱柱及流动相比例的选择
[0114]
本实施例中以出峰时间、峰形为选择标准,进行色谱柱及流动相比例的选择。
[0115]
流动相:a:乙腈(含0.1%甲酸);b:5mm甲酸铵溶液(含0.1%甲酸,5%乙腈)
[0116]
①
symmetry c18柱(2.1
×
50mm,3.5μm)
[0117]
流动相比例:90%b
[0118]
出峰时间:imp:0.38min;mer:0.48min。imp与mer在该柱无保留(图4)。
[0119]
②
atlantis t3柱(2.1
×
50mm,3μm)
[0120]
流动相比例:95%b
[0121]
出峰时间:imp:0.61min;mer:2.42min。imp在该柱无保留(图5)。
[0122]
③
hilic silica柱(2.1mm
×
50mm,3μm)
[0123]
流动相比例:15%b
[0124]
出峰时间:imp:3.68min;mer:4.64min。峰形差,且灵敏度差(图6)。
[0125]
④
hilic silica柱(2.1mm
×
50mm,3μm)
[0126]
流动相梯度:0~0.5min,15%b;0.5~2.5min,15%~40%b;2.5~3.5min,40%b;3.5~4.0min,40%~15%b;4.0~6.5min,15%b。
[0127]
出峰时间:imp:2.84min;mer:2.96min。峰形较好,灵敏度增加(图7)。
[0128]
虽然,上文中已经用一般性说明及具体实施方案对本发明作了详尽的描述,但在本发明基础上,可以对之做一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1