采用GC/LC-QTOF的土壤农药非靶向快速筛查方法
采用gc/lc
‑
qtof的土壤农药非靶向快速筛查方法
技术领域
1.本发明属于环境介质有机污染物的分析检测领域,具体涉及为一种采用gc/lc
‑
qtof的土壤农药非靶向快速筛查方法。
背景技术:
2.农药是现代农业不可或缺的一部分,可以起到防治病虫害和调节植物生长的作用。在全球范围内,每年使用近30亿公斤农药,农药行业产值约为400亿美元。在过去的30年中,我国的农药生产和使用量一直在快速增长,我国现已成为当今世界上最大的农药生产国和消费国之一。农药的广泛施用增加了农作物的产量,从而促进了粮食供应。另一方面,过量使用农药会导致有害农药残留物进入环境,例如土壤、水、植物和食物等。在土壤等复杂基质中出现大量农药残留物已成为主要环境污染问题,并且可能对健康和环境造成严重危害。要想全面认识土壤中的农药残留风险,首先需要建立土壤中合理的农药分析检测方法,主要包括提取和分析检测两个部分。
3.在提取方面,当前农药常见的提取方法包括液液萃取(liquid
‑
liquid extraction,lle)、固相萃取(solid
‑
phase extraction,spe)、基质固相分散(matrix solid
‑
phase dispersion,mspd)、固相微萃取(solid phase microextraction,spme)以及quechers提取等。其中quechers方法基于液
‑
液缓冲萃取,然后使用分散固相萃取(dspe)进行净化,并且主要允许在复杂的基质中提取回收各类极性或非极性农药。该方法因其快速有效的特点,已被广泛应用于不同类型农药的多残留分析研究中。此外,土壤基质复杂,需要在传统quechers方法的基础上,改进开发一种通用的提取方法。而在分析检测方面,色谱和质谱联合使用是最常见的分析方法,三重四极杆串联质谱由于其高选择性和灵敏度而成为最常用的联用技术,而orbitrap和tof质谱仪可以提供超高高分辨率(>20000fwhm),精确的质量测量(<5ppm),以及出色的全ms扫描灵敏度和完整的质谱信息,从而能实现目标物的定性筛选。
4.目前,最常见的农药残留研究主要是目标筛选,需要事先选择目标并并购买标准品,但覆盖面小,还会存在忽略潜在目标物的可能。因此,环境分析中最热门的趋势之一是高分辨率质谱(hr
‑
ms)与色谱(lc/gc)结合使用,在没有标准品的情况下选择合理的策略对样品中的可疑目标物进行非靶向筛查。此外,当前大多数研究都主要针对极性低,土壤结合能力弱的农药,这导致人们对土壤中半极性和极性农药的存在缺乏了解。因此,如何实现土壤基质中不同极性农药的广谱筛查是一个亟待解决的难题。
技术实现要素:
5.本发明要解决的问题是提供一种同时提取、同时净化以及广谱筛查土壤样品中农药残留的一体化方法,旨在实现土壤这种复杂基质中农药的广谱非靶向筛查,对于全面评估土壤农药污染风险有着重要意义。
6.为了解决上述技术问题,本发明提供一种采用gc/lc
‑
qtof的土壤农药非靶向快速
筛查方法,包括如下步骤:
7.(1)样品制备:
8.收集的土壤除杂混匀后进行干燥脱水(至含水率小于1%),过2mm筛,然后用研钵和研棒研磨后过100目筛的均匀粉末,作为土样(土壤样品);置于琥珀色玻璃小瓶中保存在4℃条件下备用;
9.(2)样品提取及净化:
10.将4g土样称量至50ml离心管中,加入内标使其进样含量为50ppb。静置5min后加入10ml超纯水浸泡10min,随后加入10ml含1%乙酸的乙腈后涡旋2min。添加4gmgso4和1gnacl后立即振摇1min并涡旋2min。4000rpm离心5min后将上清液转移到另一个含有300mgmgso4和50mgpsa的离心管中,手摇15s并涡旋振荡2min。最后,4000rpm离心5min后将上清液均分转移至两组氮吹管中氮吹浓缩,一组用甲醇复溶至1ml即为待测液1,过膜后进lc仪器分析,另一组用丙酮复溶至1ml为待测液2,进gc仪器分析;
11.含1%乙酸的乙腈,由1%的乙酸与99%的乙腈组成,%为体积%;
12.(3)样品检测:
13.将待测液1通过超高效液相色谱系统对农药残留化合物进行分离,采用配有电喷雾离子源的xevo tq
‑
xs,采用mrm的es+进行回收率数据采集;的采用配有电喷雾离子源的uplc
‑
q
‑
tof,分别采用ms
e
的es+和es
‑
两种模式对待测液1的前体离子和碎片离子质谱信息进行采集;
14.将待测液2通过气相色谱系统对农药残留化合物进行分离,采用配有电子轰击离子源的gc/triple quadrupole ms,采用dmrm进行回收率数据采集;采用配有电子轰击离子源的gc/q
‑
tof,采用保留时间锁定的ms1对待测液的碎片离子数据信息进行采集;
15.通过两种途径对土壤中农药残留化合物进行全面检测;
16.(4)数据分析:
17.将采集获得的待测液1数据信息利用回收率计算公式进行提取方法回收率的计算;再结合unfi软件使用unifi 1.8科学数据库对极性及中等极性农药进行筛查,根据前体离子精确质量、保留时间、碎片离子、同位素丰度以及峰强进行鉴定;
18.将采集获得的待测液2数据信息利用回收率计算公式进行提取方法回收率的计算;再结合masshunter 10.0软件使用安捷伦农药pcdl对非极性和弱极性农药进行筛查,根据保留时间、碎片离子精确质量、碎片离子共流出曲线、同位素丰度以及峰强进行鉴定;
19.快速全面分析土壤中农药残留信息。
20.所述步骤(1)中的干燥为冷冻干燥,即土壤样品干燥是在labconco真空冷冻干燥机中进行的。
21.所述步骤(3)中检测条件设置如下:
22.液相色谱条件:
23.色谱柱:acquity uplc beh c18 2.1x 100mm,1.7μm;
24.柱温:45℃;
25.流动相:a相为超纯水,b相为甲醇,二者均为hplc级,并且均加入了体积分数1%的1m乙酸铵(ph=5.0)作添加剂;
26.梯度洗脱程序:0
‑
0.25min,2%b;0.25
‑
12.25min,2%b
‑
99%b;12.25
‑
13min,维持
99%b;13
‑
13.01min,重新平衡至2%b;13.01
‑
17min,维持2%b;
27.流速:0.45ml min
‑1;
28.样品进样量:2μl;
29.esi离子源质谱条件:
30.xevo tq
‑
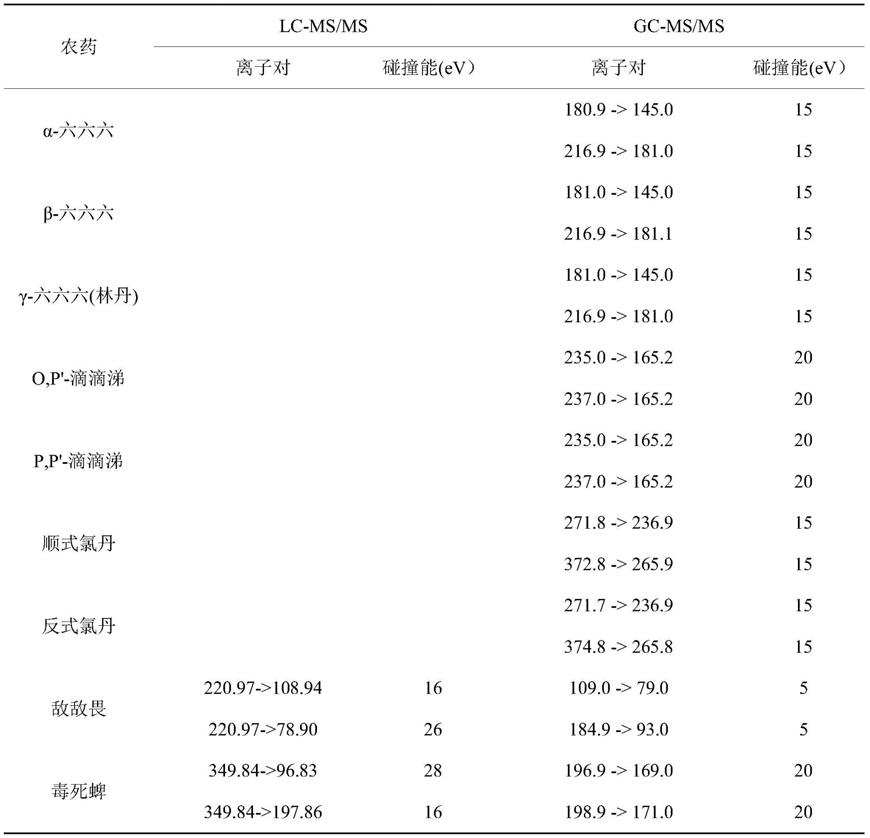
xs质谱仪,采集模式:mrm;
31.正电荷电喷雾离子源(esi+);毛细管电压为3.00kv;取样锥孔电压:20v;离子源温度150℃;脱溶剂温度:500℃;脱溶剂气体流速:800l h
‑1;锥气流量150l h
‑1;碰撞气体为0.15ml min
‑1的氩气;mrm参数见表1;
32.xevo g2
‑
xs qtof质谱仪,采集模式:ms
e
33.esi+电离模式条件:毛细管电压:1.0kv;取样锥孔电压:20v;离子源温度:120℃;脱溶剂温度:500℃;脱溶剂气体流速:900l/h;参比质量:亮氨酸脑啡呔[m+h]
+
=556.2766;
[0034]
esi
‑
电离模式条件:毛细管电压:1.0kv;取样锥孔电压:20v;离子源温度:120℃;脱溶剂温度:500℃;脱溶剂气体流速:900l/h;参比质量:亮氨酸脑啡呔[m
‑
h]
‑
=554.2615;
[0035]
采集范围:m/z 50~1000;低能碰撞能量:4ev;高能渐变碰撞能量:10~40ev。
[0036]
气相色谱条件为:
[0037]
色谱柱:2根agilent 19091s
‑
431ui hp
‑
5ms ultra inert,15m,0.25mm,0.25μm;
[0038]
升温程序:0
‑
1min 60℃;1
‑
2.5min,60℃
‑
120℃;2.5
‑
40.5min,120℃
‑
310℃;
[0039]
进样口温度:280℃;
[0040]
流速:1.05ml/min;
[0041]
载气:氮气;
[0042]
反吹条件:5min(后运行),310℃(柱温箱温度),50psi(辅助epc压力),2psi(进样口压力);
[0043]
进样量:1μl;
[0044]
ei离子源质谱条件为:
[0045]
7000d triple quadrupole ms质谱仪,采集模式:dmrm;
[0046]
传输线温度:280℃;四极杆温度:150℃;离子源温度:300℃;电子能量:70ev;dmrm具体参数见表1;
[0047]
7250q
‑
tof质谱仪,采集模式:ms1
[0048]
传输线温度:280℃;四极杆温度:150℃;离子源温度:300℃;电子能量:70ev;谱图采集速率:4hz;质量数范围:m/z 35
‑
1000;
[0049]
所述步骤(4)中,unifi方法检索参数为:保留时间容差
±
0.2min,精确质量匹配容差为
±
5mda,离子化形式选择+h,+na和
‑
h模式;masshunter方法检索参数为:保留时间容差
±
0.2min,精确质量匹配容差:
±
20ppm。
[0050]
在本发明中:
[0051]
1、通过优化土壤农药提取净化方法,同时综合考虑回收效率、时间、耗材及人力成本,本发明最终乙腈作为提取剂,psa作为净化剂的quechers方法作为同时提取土壤中农药残留的方法;
[0052]
2、通过结合使用气相色谱和液相色谱,并基于非靶向筛查方法,优化筛查方法,最终建立起一种采用gc/lc
‑
qtof的土壤农药非靶向快速筛查方法。
[0053]
与现有技术相比,本发明的优点和有益效果是:
[0054]
1、选择quechers提取作为前处理方法,能有效去除土壤样品中的基质干扰物质,农药残留的回收率可接受,方便快捷;
[0055]
2、结合气相色谱和液相色谱,对不同极性的农药均可分析检测,可实现样品中农药的广谱检测。并选择tof作为质量检测器,具有较高分辨率和灵敏度,可为后续数据分析提供全面精确的色谱质谱信息;
[0056]
综上所述,本发明方法快速,且可以实现高通量广谱的农药残留分析,可为土壤中农药残留的快速筛查和风险管控提供重要的方法依据和数据支持。
附图说明
[0057]
下面结合附图对本发明的具体实施方式作进一步详细说明。
[0058]
图1是实际样品在液相上的总离子流图;
[0059]
图2是实际样品在液相上的总离子流图;
[0060]
图3是实际样品中典型农药三环唑在液相上的提取离子流图;
[0061]
图4是实际样品中典型农药三环唑在unifi软件上的鉴定示例;结合图3显示的液相色谱提取离子色谱峰形及保留时间和图4中的精确质量偏差以及碎片离子等可实现三环唑这种极性农药的身份鉴定;
[0062]
图4中,上图为低能量通道中农药三环唑的质谱图,下图为高能量通道中农药三环唑的质谱图;
[0063]
图5是实际样品中典型农药α
‑
六六六在气相上的提取离子流图;
[0064]
图6是实际样品中典型农药α
‑
六六六在masshunter上的鉴定示例,结合图5显示的液相色谱提取离子色谱峰形及保留时间和图6中的精确质量偏差以及共流出曲线等可实现α
‑
六六六这种弱极性农药的身份鉴定。
[0065]
图6中,左上为农药α
‑
六六六各碎片离子的提取离子流图,左下为农药α
‑
六六六各碎片离子的共流出曲线,右上为农药α
‑
六六六软件自动净化后的质谱图,,右下为农药α
‑
六六六的实际质谱图。
具体实施方式
[0066]
下面结合具体实施例对本发明进行进一步描述,但本发明的保护范围并不仅限于此:
[0067]
实施例1:
[0068]
一、试剂及药品
[0069]
除非另有说明,分析时所使用的试剂均为符合国家标准的色谱级。实验用水是mili
‑
q超纯水。
[0070]
丙酮(c3h6o):色谱纯;
[0071]
甲醇(ch3oh):色谱纯;
[0072]
乙腈(c2h3n):色谱纯;
[0073]
乙酸(ch3cooh):色谱纯;
[0074]
乙腈酸性混合溶剂:乙腈中加入体积1%乙酸混合;
[0075]
农药单标储备液:100μg ml
‑1(每种农药),溶于甲醇,
‑
20℃保存。
[0076]
农药混标中间溶液:5μg ml
‑1(每种农药),溶于丙酮,
‑
20℃保存。
[0077]
内标中间溶液:5μg ml
‑1(每种内标),溶于丙酮,
‑
20℃保存。
[0078]
准确称取适量的农药混标中间溶液以及进样内标中间溶液分别使用甲醇和丙酮制备成含24种农药混合浓度为1ng ml
‑1、5ng ml
‑1、10ng ml
‑1、50ng ml
‑1、100ng ml
‑1、200ng ml
‑1和含进样内标物浓度为50ng ml
‑1的混合标准工作溶液,
‑
20℃保存。
[0079]
农药具体为如表1所述的23种农药;
[0080]
内标具体为:p,p'
‑
滴滴涕
‑
d8、毒死蜱
‑
d10、氟虫腈
‑
13c415n2、吡虫啉
‑
d4、阿特拉津
‑
d5。
[0081]
二、仪器及设备
[0082]
超高效液相色谱
‑
三重四级杆质谱仪(waters acquity uplc i
‑
class/xevo tq
‑
xs);超高效液相色谱
‑
飞行时间质谱仪(waters acquity uplc i
‑
class/xevo g2
‑
xs qtof)acquity uplc beh c18色谱柱(2.1mm
×
100mm,1.7μm);气相色谱
‑
三重四级杆质谱仪(agilent 7010b gc/7000d triple quadrupole ms);气相色谱
‑
飞行时间质谱仪(agilent 8890gc/7250q
‑
tof);organomation氮吹仪;一般实验室常用仪器和设备。
[0083]
三、所述采用gc/lc
‑
qtof的土壤农药非靶向快速筛查方法包括如下步骤:
[0084]
1、样品制备
[0085]
采集的土壤样品置于棕色玻璃瓶中,玻璃瓶先用超纯水洗净,于马弗炉中450℃灼烧10h以彻底去除玻璃瓶内残留的有机物等;采集后的土壤样品于4℃以下冷藏、避光、密封保存,尽快带到实验室分析。
[0086]
将土壤样品放在搪瓷盘上,混匀,除去枝棒、叶片、石子等异物,放于真空冷冻干燥机中干燥脱水(0.2mbar的真空度、
‑
50℃,干燥5d,此时样品的至含水率小于1%);干燥后的土壤过2mm筛,然后用研钵和研棒研磨至过100目筛的均匀粉末,置于琥珀色玻璃小瓶中保存在4℃条件下备用。
[0087]
2、样品前处理
[0088]
因为常用的提取方法后需要经固相萃取柱进行净化除杂,该过程通常耗时耗力。因此在实验室现有条件下,本发明设定使用quechers提取对样品进行前处理操作。
[0089]
具体步骤为:将4g步骤1所得的土样称量至50ml离心管中,加入内标使内标进样含量为50ppb。静置5min后加入10ml超纯水浸泡10min,随后加入10ml含1%乙酸的乙腈后涡旋2min。添加4gmgso4和1gnacl后立即振摇1min并涡旋2min。4000rpm离心5min后将上清液转移到另一个含有300mgmgso4和50mgpsa的离心管中,手摇15s并涡旋振荡2min。最后,4000rpm离心5min后将上清液均分转移至两组氮吹管中氮吹浓缩,一组用甲醇复溶至1ml即为待测液1,过膜后进lc仪器分析,另一组用丙酮复溶至1ml为待测液2,进gc仪器分析。
[0090]
3、样品测定
[0091]
(1)液相色谱条件:
[0092]
色谱柱:acquity uplc beh c18(2.1mm
×
100mm,1.7μm);柱温45℃;样品进样量为2μl;
[0093]
流动相:a相为超纯水,b相为甲醇,二者均为hplc级,并且均加入了体积分数1%的1m乙酸铵(ph=5.0)作添加剂;流速为0.45ml min
‑1;
[0094]
a相为:99%超纯水与1%的1m乙酸铵溶液的混合液,%为体积%;
[0095]
b相为:99%甲醇与1%的1m乙酸铵溶液的混合液,%为体积%。
[0096]
流动相的洗脱梯度为:0
‑
0.25min,2%b;0.25
‑
12.25min,2%b
‑
99%b;12.25
‑
13min,维持99%b;13
‑
13.01min,重新平衡至2%b;13.01
‑
17min,维持2%b;
[0097]
(2)xevo tq
‑
xs质谱条件:
[0098]
采集模式:mrm;
[0099]
正电荷电喷雾离子源(esi+);毛细管电压为3.00kv;取样锥孔电压:20v;离子源温度150℃;脱溶剂温度:500℃;脱溶剂气体流速:800l h
‑1;锥气流量150l h
‑1;碰撞气体为0.15ml min
‑1的氩气;mrm参数见表1;
[0100]
说明:mrm的参数是为了量化上述前处理方法的回收率,从而确定合适的前处理方法。
[0101]
(3)xevo g2
‑
xs qtof质谱条件:
[0102]
采集模式:ms
e
[0103]
正电荷电喷雾离子源(esi+);毛细管电压:1.0kv;取样锥孔电压:20v;离子源温度:120℃;脱溶剂温度:500℃;脱溶剂气体流速:900l/h;参比质量:亮氨酸脑啡呔[m+h]
+
=556.2766;
[0104]
负电荷电喷雾离子源(esi
‑
);毛细管电压:1.0kv;取样锥孔电压:20v;离子源温度:120℃;脱溶剂温度:500℃;脱溶剂气体流速:900l/h;参比质量:亮氨酸脑啡呔[m
‑
h]
‑
=554.2615;
[0105]
采集范围:m/z 50~1000;低能碰撞能量:4ev;高能渐变碰撞能量:10~40ev。
[0106]
(4)气相色谱条件:
[0107]
色谱柱:2根agilent 19091s
‑
431ui hp
‑
5ms ultra inert(15m,0.25mm,0.25μm);进样口温度:280℃;流速:1.05ml/min;载气:氮气;反吹条件:5min(后运行),310℃(柱温箱温度),50psi(辅助epc压力),2psi(进样口压力);进样量:1μl;
[0108]
(5)7000d triple quadrupole ms质谱条件:
[0109]
采集模式:dmrm
[0110]
传输线温度:280℃;四极杆温度:150℃;离子源温度:300℃;电子能量:70ev;dmrm具体参数见表1;
[0111]
(6)7250q
‑
tof质谱条件:
[0112]
采集模式:ms1
[0113]
传输线温度:280℃;四极杆温度:150℃;离子源温度:300℃;电子能量:70ev;谱图采集速率:4hz;采集范围:m/z 35
‑
1000。
[0114]
4、筛查方法:
[0115]
按照上述步骤3,将待测液1通过超高效液相色谱系统对农药残留化合物进行分离,采用配有电喷雾离子源的xevo tq
‑
xs,根据上述(1)、(2)条件对步骤1所述的农药混标进行回收率测定;采用配有电喷雾离子源的uplc
‑
q
‑
tof,分别采用ms
e
的es+和es
‑
两种模式对待测液1的前体离子和碎片离子质谱信息进行采集,即,根据上述(1)条件可得到液相色谱上的保留时间数据,根据上述(3)条件可得到前体离子精确质量和碎片离子等数据,将采集获得的待测液1数据信息结合unfi软件使用unifi 1.8科学数据库对极性及中等极性农
药进行筛查,根据上述(1)、(3)条件下得到的前体离子精确质量、保留时间、碎片离子、同位素丰度以及峰强等信息进行鉴定;
[0116]
按照上述步骤3,将待测液2通过气相色谱系统对农药残留化合物进行分离,采用配有电子轰击离子源的gc/triple quadrupole ms,根据上述(4)、(5)条件对步骤1所述的农药混标进行回收率测定;采用配有电子轰击离子源的gc/q
‑
tof,采用保留时间锁定的ms1对待测液的碎片离子数据信息进行采集,即,根据上述(4)条件可得到气相色谱上的保留时间数据,根据上述(6)条件可得到碎片离子精确质量和碎片离子共流出曲线等数据,将采集获得的待测液2数据信息结合masshunter 10.0软件使用安捷伦农药pcdl对非极性和弱极性农药进行筛查,根据上述(4)、(6)条件下得到保留时间、碎片离子精确质量、碎片离子共流出曲线、同位素丰度以及峰强进行鉴定,快速全面分析土壤中农药残留信息。
[0117]
即,通过两种途径对土壤中农药残留化合物进行全面检测。
[0118]
四、结果与讨论
[0119]
1、土壤样品前处理方法确定
[0120]
土壤样品基质复杂,存在着大量的基质干扰物,因此需要选择合适的前处理方法以实现土壤中农药的全面提取。本实验首先选择了选定了23种官能团各异、极性不同、类型不同的农药作为代表,这些农药用途各异,理化性质覆盖广泛,可作为代表性物质用作土壤前处理方法的开发,根据上述步骤3中的(2)条件及(5)条件确定了其lc
‑
ms/ms和gc
‑
ms/ms质谱参数(表1)。这24种农药涵盖了杀虫剂、除草剂、杀菌剂等最常用农药,在杀虫剂中包含了有机氯、有机磷、菊酯类、新烟碱类等目前正在广泛使用和在历史上曾经广泛生产使用的品种。同时还选择了p,p'
‑
滴滴涕
‑
d8、毒死蜱
‑
d10、氟虫腈
‑
13
c4
15
n2、吡虫啉
‑
d4、阿特拉津
‑
d5这5种同位素标记化合作为内标以便精确定量。
[0121]
表1、23种代表性农药及其同位素标记化合物的主要质谱参数
[0122]
[0123]
[0124][0125]
并以这23种农药为代表,开发验证了能够快速、方便提取众多性质各异农药的提取方法。根据上述步骤3中的(1)、(2)条件及(4)、(5)条件确定的具体回收率见表2。回收率计算公式为:k=(a
‑
b)/c
×
100%,式中,a:加入标准物质的样品测得量;b:样品中该物质的测得量;c:加入的标准物质量。
[0126]
根据表2,净化剂为psa组内所有农药的回收率均在60%
‑
140%之间,可实现样品中目标物的充分提取。此外毒死蜱、高效氯氰菊酯、丁草胺等15种农药可以在gc
‑
ms/ms和lc
‑
ms/ms同时检出,交叉验证,极大得增加了方法的可靠性。
[0127]
表2典型农药回收率
[0128]
[0129][0130]
2、土壤样品前处理方法确定
[0131]
unifi方法检索参数为:保留时间容差
±
0.2min,精确质量匹配容差为
±
5mda,离子化形式选择+h,+n和
‑
h模式;masshunter方法检索参数为:保留时间容差
±
0.2min,精确质量匹配容差:
±
20ppm。
[0132]
3、非靶向快速筛查质控方法
[0133]
23种质控农药以50ppb的浓度加标于基质样品中,并在上述步骤1、2及步骤3中的条件(1)、(3)、(4)、(6)下进行实验,液相检出结果见表3,气相检出结果见表4;加标农药均被检出,可说明本筛查方法适用范围广泛,可应用于实际土壤样品。
[0134]
表3、质控样品液相检出结果
[0135]
[0136][0137]
表4质控样品气相检出结果
[0138]
[0139][0140]
4、非靶向快速筛查农药残留的实际应用
[0141]
将上述筛查方法应用于实际土壤样品,上述步骤1、2及步骤3中的条件(1)、(3)、(4)、(6)下进行实验,总离子流图见图1和图2。实际样品筛查结果见表5,在该实际土壤样品中共检出农药96种,涵盖有机氯、有机磷、氨基甲酸酯、拟除虫菊酯等不同类别的农药,在液相和气相检出的代表性农药的鉴定示例见图3
‑
6。其中仅在液相上检出的农药有41种,仅在气相上检出的农药有31种,在液相和气相上均有检出的有24种,由此也说明了将气相色谱和液相色谱联合使用能极大的拓展方法的分析检测范围,本方法也能实现土壤样品种高通量广谱的农药残留非靶向筛查。
[0142]
表5实际样品筛查结果
[0143]
[0144]
[0145]
[0146]
[0147][0148]
对比例1、将实施例1“样品前处理”中的提取方法,将净化剂由“c18”改为“psa”,其余等同于实施例1。
[0149]
所得结果如表2所述,根据表2,当净化剂为c18时,α
‑
六六六、p,p'
‑
滴滴涕和丁草胺等农药回收率均不满足要求,而当净化剂为psa时,所有农药的回收率均在60%
‑
140%之间,可实现样品中目标物的充分提取,因此选择添加psa作为净化剂的quechers方法作为样品前处理方法。
[0150]
对比例2、将实施例1样品测定步骤中采用的“液相色谱”改为“液相色谱于气相色谱联合使用”,其余等同于实施例1。
[0151]
所得结果如表3及表4所述:若仅单独使用液相色谱,则仅能实现强极性和中等极性农药的筛查与检测,而将液相色谱与气相色谱联合使用,则可以实现包括有机氯农药等在内的弱极性农药的检测,可以大大拓宽应用范围。因此,选择将液相色谱与气相色谱联合使用。
[0152]
最后,还需要注意的是,以上列举的仅是本发明的若干个具体实施例。显然,本发明不限于以上实施例,还可以有许多变形。本领域的普通技术人员能从本发明公开的内容直接导出或联想到的所有变形,均应认为是本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1