一种固相萃取包与检测方法与流程
本发明属于分析检测技术领域,具体涉及一种固相萃取包与检测方法。
背景技术:
果酒因其酒精度低、绿色健康、色彩缤纷和变幻口感广受年轻消费群体等新酒民的欢迎,市场上常见的是配制型果酒,即以新鲜营养的水果为原料,利用自然发酵或人工添加酵母菌制造,在工艺上融合了各种颜色水果和低浓度酒精的双重风味,增添了味蕾体验。近年来,随着饮酒文化的变迁,一些不法商贩为了刺激消费、快速掘金,在果酒中违法添加各种合成色素以增加果酒的感官效应,造成了大量表面颜色鲜亮、实则是掺伪勾兑的假果酒流向市场,果酒中色素超标、超范围使用,以及违禁使用不合格或禁用色素的现象屡禁不止,引发食品安全问题事件,给消费者造成了财产损失和健康隐患。
色素分为天然色素和人工合成色素两种,天然色素由于成本高昂、着色力差,很少在食品加工中使用;人工合成色素多为色泽鲜亮、稳定性好、廉价易得,因而广泛使用。但是,人工合成色素大多以苯及苯系物等化工产品经有机反应制得偶氮类或芳基甲烷类化合物,具有致敏性和致癌性。我国国家标准gb2760-2014《食品安全国家标准食品添加剂使用标准》准许使用的人工合成色素仅有10种,同时对配制酒中人工色素的允许使用品种、范围和添加量做出了严格限量。
但目前国标仅仅对配制酒中的柠檬黄、新红、苋菜红、胭脂红、日落黄、亮蓝和赤鲜红等7种酸性色素采用聚酰胺粉末吸附法等前处理方法并进行高效液相色谱(hplc)法测定(食品安全国家标准食品中合成着色剂的测定gb5009.35-2016),也有研究者对葡萄酒中少数几种人工色素进行了固相萃取-hplc测定(文献1:王浩等.固相萃取-hplc法同时测定葡萄酒中8种人工色素.中国食品添加剂,2010(4):281-285.文献2:李小平等.固相萃取-反相高效液相色谱法同时测定葡萄酒中7种合成色素,中国卫生检验杂志,2013,23(12):2566-2569.)。以上几种方法针对待测目标物的检测种类较少,一般仅对几种常见传统合成色素建立了相关检验标准和操作方法,而对新型食用合成色素的检验检测试验数据则相对缺乏,无法满足当前市场监管与检验机构的检测需求;样品前处理方法中,吸附过滤法耗时费力、试剂回收性差,固相萃取法在上样前必须使用大量有机溶剂进行预洗活化和淋洗弃液过程,溶剂用量大、损耗多、费时长且商品化的柱体昂贵。目前针对少数几种目标物色素的传统检测方法远远不能满足实验室检测的要求。研究热点更多的集中在如何有效去除果酒样品中复杂基质对多组分人工合成色素定性和定量分析的干扰,以及如何实现多组分人工合成色素在色谱柱上的基线分离这两个问题。面对市场上非法制造的各类果酒产品,为了提高工作效率、降低实验成本、提升工作质量,亟需建立多种人工合成色素简单快速前处理与同时在线定量检测技术以满足现场与实验室全面、快速、精准、高效检测的要求。
技术实现要素:
为了解决现有技术中存在的上述问题,本发明旨在提供本发明可用于果酒中色素吸附解析前处理的混合固相萃取包,用于去除果酒中的复杂基质,取得了良好的富集和净化效果。
本发明的另一个目的是提供一种利用hplc法对合成色素进行快速定量检测的方法。
为了实现上述发明目的,本发明提供如下技术方案:
一种固相萃取吸附包,其包括固相萃取盐析包和固相萃取吸附材料,所述固相萃取盐析包和固相萃取吸附材料独立包装,其中所述固相萃取吸附材料为聚苯胺负载聚苯乙烯微球,所述聚苯胺负载聚苯乙烯微球粒径大小为110±5nm,且其红外图谱在1400cm-1,1642cm-1,3457cm-1处出现特征峰。
优选地,所述固相萃取盐析包中为氯化钠和无水硫酸钠的混合物,二者质量比为1:1-1:6。
本发明提供的聚苯胺负载聚苯乙烯微球表面本身具有丰富的含氧基团和优良吸附特性,配合无水硫酸钠既可以作为脱水干燥剂也可以作为盐析剂,无水硫酸钠与氯化钠混合使用效果更佳。
本发明所述的聚苯胺负载聚苯乙烯微球可以反复使用,所述微球的回收方法为:盐析反应完成后的沉积产物依次先后用无水乙醇、2mol/l稀盐酸和0.5%氨水反复离心洗涤数次,3000-5000rpm转速离心5-10min,用二次去离子水洗至溶液ph7。产品在50-80℃烘箱烘干24h,即可反复使用3-5次,萃取效率维持在71%-92%。
本发明提供了一种低温模板聚合法合成了所述新型固相萃取吸附材料聚苯胺负载聚苯乙烯微球:首先使用非乳化一锅法制备聚苯乙烯微球(ps),并以此为模板,经磺化后利用低温聚合法负载聚苯胺纳米颗粒(pani),实现了pani包覆ps(pani@ps)的材料合成。
优选地,所述聚苯胺负载聚苯乙烯微球的合成方法,包括如下步骤:
(1)聚苯乙烯微球ps的制备
将苯乙烯单体和水的混合物超声分散后,加入过硫酸钾水溶液,惰性环境下发生聚合反应,反应结束后水洗、离心、干燥,得ps微球;
(2)聚苯乙烯微球的磺化
将步骤(1)所得ps微球置于浓硫酸中,超声分散均匀,30-50℃恒温反应0.5-2小时,离心分离,对所得固体进行冷冻干燥,得到磺化的ps微球;
(3)聚苯胺负载聚苯乙烯微球的制备
将步骤(2)所得磺化的ps微球和磷酸水溶液低温超声分散均匀,加入适量苯胺在冰浴条件下搅拌,同时逐滴加入过硫酸铵溶液,滴加完成后继续冰浴搅拌8-12h,反应完成后,产物依次用无水乙醇和稀盐酸反复离心洗涤,离心,用二次去离子水洗至溶液ph=7,稀氨水搅拌脱氢处理,干燥,得聚苯胺颗粒负载聚苯乙烯微球。
优选地,步骤(1)中聚合反应的温度为70-90℃;和/或聚合反应的时间为5-5h;进一步优选为5-8h。
优选地,步骤(1)中所述苯乙烯单体是以溶液的形式加入,其体积分数为4%-6%。
优选地,步骤(1)中过硫酸钾水溶液的浓度为0.5-1.0mol/l。
优选地,步骤(1)中所述苯乙烯单体在进行步骤(1)前进行如下预处理:采用质量百分比为5%-15%氢氧化钠溶液进行碱洗,二次去离子水洗涤多次,直到水溶液呈中性。
优选地,步骤(1)所述ps微球的粒径大小为100nm±10nm。
优选地,步骤(2)中ps微球与浓硫酸的质量体积比为1:1-1:20。
优选地,步骤(3)中所述磷酸水溶液的浓度为1-3mol/l。
优选地,步骤(3)中苯胺与磺化ps微球的质量比为1:1-3:1。
优选地,步骤(3)中苯胺是以溶液的形式加入,其浓度为1.0-3.0mol/l。
优选地,步骤(3)中干燥的温度为50-80℃;和/或干燥的时间为8-12h。
优选地,步骤(3)中所述过硫酸铵溶液的浓度为2mol/l。
优选地,步骤(3)中所述过硫酸铵溶液与磺化的ps微球的质量比为1:1-1:1.2。
聚苯乙烯微球是一个良好的介质材料,既可以用做合成材料的模板,也可以通过改性成为功能化载体。聚苯胺是一种常见的高分子化合物,易于合成、原料易得、用途广泛。针对复杂吸附环境,聚苯胺耐溶剂性能好、适应能力强,加上改性后的聚苯胺纳米材料表面含有大量的含氧基和氨基功能化基团,在吸附环境中的重金属、有机染料等方面应用广泛。
其原理为:水溶性聚苯胺@聚苯乙烯复合材料本身具有吸附水溶性色素和杂质的特点,这是由其物理性质(比表面积和颗粒结构)和化学性质(表面化学、极性和非极性相互作用力)共同决定的。根据相似相溶原理,材料更加倾向于亲水性,水溶液中的溶剂更易发生水-聚苯胺液固界面扩散作用;复合材料本身遵循化学平衡放热原理,有机溶剂同时参与竞争复合材料的固相吸附成分更有利于固相净化萃取过程;另外,由于复合材料的表面官能团类别和数量较多,如羟基、羧基和磺化基团等含氧官能团,它们对色素杂质的吸附具有促进作用。
本发明还提供了所述固相萃取吸附包在对果酒中色素含量的检测中的应用,其包括如下步骤:
样品预处理步骤:
1)向果酒中加入碎瓷片或沸石,去除乙醇,得提取液;
2)加入2.0-5.0ml乙醇-1%柠檬酸涡旋至步骤1)所得提取液混合均匀;
3)向2)加入无水硫酸钠/氯化钠混合固相萃取包,震荡提取10-20min,离心5-10min,移取上清液;
4)向3)加入0.20-0.40g聚苯胺负载聚苯乙烯微球涡旋,混匀离心,移取上清液,沉积物另存待洗;
5)上述4)溶液40℃-70℃氮吹浓缩;用乙腈-0.1%氨水复溶,过滤膜,上机待测;
样品检测步骤:
色谱分析采用安捷伦高效液相色谱仪串联紫外可见二极管阵列检测器,色谱柱为dikmaodsc18色谱柱,4.6mm×250mm,5μm;柱温30-40℃;流动相:a/b=0.05%乙酸铵/乙腈;流速1.0ml/min;进样体积20μl;采用梯度洗脱进样分离;hplc梯度洗脱条件为:流动相a/b=0.05%乙酸铵/乙腈,0-5min:a/b=5%/95%;5/10min:a/b=10%/90%;10-16min:a/b=25%/75%;16-20min:a/b=40%/60%;20-30min:a/b=55%/45%;30-40min:a/b=85%/15%;40-50min:a/b=5%/95%;二极管阵列检测器波长设置为254nm。
在分析样品组之前,注入空白溶剂乙腈以确保系统没有污染物或干扰峰。
与现有技术相比,本发明提供的混合固相萃取包用于去除果酒中的复杂基质,取得了良好的富集和净化效果。该固相萃取包由(1)聚苯胺负载聚苯乙烯微球,(2)无水硫酸钠和氯化钠固体按一定质量比混合而成。通过多次解吸液进行循环洗脱后还可以反复使用。
本发明建立了一种高效液相色谱联合紫外检测器的21种合成色素的快速定量检测方法。该方法造价低廉、快速高效、重复性好、准确度高,能够弥补现有标准检测方法和测试品种的局限。
与现有食品安全国家标准方法(gb5009.35-2016)比较,在原有7种色素的基础上,新增了14种可检测的人工合成色素,适用范围广。
附图说明
图1为本发明提供的ps微球的扫描电镜图;
图2为本发明提供的磺化ps微球的扫描电镜图;
图3为本发明提供的pani@ps微球的扫描电镜图;
图4为本发明提供的pani@ps微球的红外图谱;
图5为21种色素的20μg/ml混合标准溶液的hplc检测图谱。
图6为20μg/ml的柠檬黄标准溶液的hplc检测图谱;
图7为20μg/ml的新红标准溶液的hplc检测图谱;
图8为20μg/ml的胭脂红标准溶液的hplc检测图谱;
图9为20μg/ml的苋菜红标准溶液的hplc检测图谱;
图10为20μg/ml的日落黄标准溶液的hplc检测图谱;
图11为20μg/ml的酸性红2g标准溶液的hplc检测图谱;
图12为20μg/ml的溴酚蓝标准溶液的hplc检测图谱;
图13为20μg/ml的曙红钠标准溶液的hplc检测图谱;
图14为20μg/ml的亮蓝标准溶液的hplc检测图谱;
图15为20μg/ml的赤藓红标准溶液的hplc检测图谱;
图16为20μg/ml的酸性橙ⅱ标准溶液的hplc检测图谱;
图17为20μg/ml的漆黄素标准溶液的hplc检测图谱;
图18为20μg/ml的甲基红标准溶液的hplc检测图谱;
图19为20μg/ml的偶氮玉红标准溶液的hplc检测图谱;
图20为20μg/ml的亚甲基蓝标准溶液的hplc检测图谱;
图21为20μg/ml金胺(碱性嫩黄o)标准溶液的hplc检测图谱;
图22为20μg/ml碱性橙2标准溶液的hplc检测图谱;
图23为20μg/ml罗丹明b标准溶液的hplc检测图谱;
图24为20μg/ml碱性橙g标准溶液的hplc检测图谱;
图25为20μg/ml碱性橙r标准溶液的hplc检测图谱;
图26为20μg/ml角黄素标准溶液的hplc检测图谱;
图27为青梅酒样品的hplc检测图谱;
图28为百香果酒样品的hplc检测图谱;
其中,图4中,从1-3分别代表ps微球;磺化ps微球;pani@ps复合纳米球。
具体实施方式
为了使本发明实现的技术手段、创作特征、达成目的与功效易于明白了解,下面结合具体实施例,进一步阐明本发明,但下述实施例仅仅为本发明的优选实施例,并非全部。基于实施方式中的实施例,本领域技术人员在没有做出创造性劳动的前提下所获得其它实施例,都属于本发明的保护范围。
下述实施例中,若无特殊说明,所用的操作方法均为常规操作方法,所用设备均为常规设备。
实施例1
(1)聚苯乙烯微球(ps)的制备方法:
引发剂:苯乙烯(st)单体,化学纯,使用前进行减压蒸馏提纯;
苯乙烯单体的减压蒸馏提纯方法:氢氧化钠naoh质量百分比5%进行碱洗,二次去离子水洗涤多次,直到水溶液呈中性。
过硫酸钾(k2s2o8,kps),分析纯,使用前重结晶提纯精制。
制备方法:100ml三口烧瓶中加入15ml6%苯乙烯单体和30ml水,室温超声分散20min,一次性加入浓度为0.5mol/l的kps水溶液。通氮保护下恒温90℃聚合反应6h,水洗离心烘干,合成ps球粒径大小在100nm。
(2)聚苯乙烯微球的磺化方法:
将5.0g的ps微球置于50mll体积的浓硫酸中,超声分散10min,40℃水浴1小时,冷冻干燥得到磺化的ps球。
(3)聚苯胺负载聚苯乙烯微球的方法:低温氧化聚合法
在250ml三口瓶中加入10.0g磺化ps球和70ml2mol/l磷酸水溶液低温超声分散均匀,加入1.0mol/l苯胺(苯胺与磺化ps微球的质量比为1:1)在冰浴条件下搅拌20min,磁子搅拌机的搅拌速度维持在200-500rpm,逐滴加入2mol/l过硫酸铵,其中过硫酸铵与苯胺的物料比1:1.2的,滴速3-5秒/滴,继续冰浴搅拌8-12h,反应完成后,产物依次用无水乙醇和2mol/l稀盐酸反复离心洗涤,3000-5000rpm转速离心5-10min,用二次去离子水洗至溶液ph7。按照1g/100ml稀氨水搅拌1h脱氢处理。产品在50-80℃烘箱烘干8-12h,聚苯胺颗粒负载聚苯乙烯微球粒径110±5nm,产物呈墨绿色。
如图1-3所示,为步骤(1)-(3)依次制得的ps微球、磺化ps微球和pani@ps复合纳米微球的扫描电镜图sem。从图中可以看出,ps微球粒径大小均匀、分散性好,溶于dmf后的扫描电镜表明,其粒径大小保持在在100nm左右;经硫酸磺化的ps微球呈现黏连状,可能与其磺化性质有关;pani复合纳米球均匀包覆在ps上,形成了粒径大小110nm左右的pani@ps复合纳米球。
如图4所示,为步骤(1)-(3)依次制得的ps微球、磺化ps微球和pani@ps复合纳米微球的红外对比图谱,从1-3分别代表ps微球;磺化ps微球;pani@ps复合纳米球。ps球苯基在1492cm-1,1451cm-1,756cm-1,698cm-1出现特征振动峰,亚甲基在2926cm-1和2851cm-1出现伸缩振动吸收峰;
磺化ps球的-so3h基团振动峰出现在1236cm-1处,表明磺酸根离子的存在;pani在1400cm-1,1642cm-1,3457cm-1位置分别为聚苯胺的c=c,nh3+,-oh伸缩振动特征峰,表明pani@ps的成功合成。
(4)混合萃取包的配制
萃取包1:1.0g:1.0g-1.0g:6.0g的氯化钠和无水硫酸钠(质量百分相比1:1-1:6),为固相萃取盐析包;
萃取包2:pani@ps0.20-0.40g,为固相萃取吸附材料;
萃取包1和2独立包装,按先后顺序进行前处理使用。
实施例2
果酒中色素含量的检测
1样品前处理
步骤1):称取10g样品于50ml玻璃烧杯或圆底烧瓶中,加入碎瓷片或沸石,去除乙醇;
步骤2):加入5.0ml乙醇-1%柠檬酸涡旋1-2min直至提取液混合均匀;
步骤3):向2)加入无水硫酸钠/氯化钠混合固相萃取包,震荡提取10-20min,3000-5000rpm离心5-10min,移取上清液;
步骤4:向3)加入0.20gpani@ps涡旋1-2min,混匀离心(转速3000-5000rpm,时间5-10min),移取上清液,沉积物另存待洗;
步骤5):上述4)溶液置于氮吹仪中60℃氮吹浓缩;用1.0ml乙腈-0.1%氨水复溶,过0.22μm滤膜,上机待测。
2检测方法的建立
2.1标准液配制
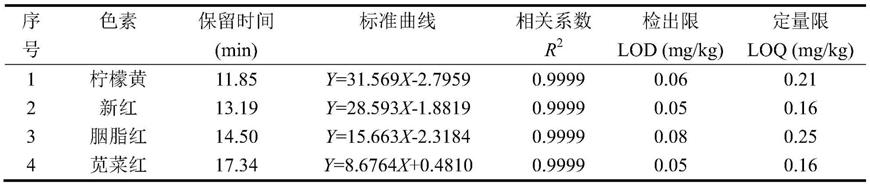
1)通过系列稀释,将质量浓度20μg/ml的基质匹配混合工作液逐级稀释后分别进样测定,以质量浓度为横坐标,峰面积为纵坐标,建立21种色素的基质校正标准曲线,各色素的标准曲线及检出限情况如下表1所示。
表121种色素的保留时间、标准曲线、相关系数、检出限和定量限
从表1可以看出,利用本发明建立的hplc检测方法的检出限低于0.16mg/kg,而果酒中色素的限量一般为0.025-0.10g/kg,所以,可以满足绝大部分实验的需求。
2)分别配制质量浓度为0.1,0.2,0.5,1.0,2.0,5.0,10.0,20.0μg/ml的混合标准溶液,绘制标准曲线,结果表明,21种人工合成色素在0.4μg/ml-20.0μg/ml质量浓度范围内呈现良好线性关系,r2≥0.9997。如图5所示为21种色素的20.0μg/ml混合标准溶液的hplc检测图谱。图6-26为21种色素单一成分标准溶液的hplc检测图谱。
图5中1-21号出峰位置分别代表:
1.柠檬黄;2.新红;3.胭脂红;4.苋菜红;5.日落黄;6.酸性红2g;7.溴酚蓝;8.曙红钠;9.亮蓝;10.赤藓红;11.酸性橙ⅱ;12.漆黄素;13.甲基红;14.偶氮玉红;15.亚甲基蓝;16.金胺(碱性嫩黄o);17.碱性橙2;18.罗丹明b;19.碱性橙g;20.碱性橙r;21.角黄素。
2.2样品中色素的含量检测
本实施例对市售30种果酒种类和12种葡萄酒样品进行测定:
果酒:30种,菠萝酒、薄荷酒、百香果酒、草莓酒、哈密瓜酒、橘子酒、金桔酒、梨子酒、荔枝酒、蓝莓酒、龙眼酒、榴莲酒、木瓜酒、芒果酒、猕猴桃酒、蜜桃酒、柠檬酒、苹果酒、枇杷酒、青梅酒、桑椹酒、山楂酒、石榴酒、柿子酒、西瓜酒、柚子酒、杨梅酒、椰子酒、樱桃酒、无花果酒。各选2种市售不同品牌,样品数量60份。
葡萄酒:12种市售不同品牌,样品数量12份。
总计样品数量72份。
2.2.1检测条件:
色谱分析采用安捷伦高效液相色谱仪串联紫外可见二极管阵列检测器,色谱柱为dikmaodsc18色谱柱(4.6mm×250mm,5μm);柱温30-40℃;流动相:0.05%乙酸铵(a)-乙腈(b);流速1.0ml/min;进样体积20μl;采用梯度洗脱进样分离。hplc梯度洗脱条件为:流动相a/b(0.05%乙酸铵/乙腈),0-5min:a/b=5%/95%;5/10min:a/b=10%/90%;10-16min:a/b=25%/75%;16-20min:a/b=40%/60%;20-30min:a/b=55%/45%;30-40min:a/b=85%/15%;40-50min:a/b=5%/95%;二极管阵列检测器波长设置为254nm。在分析样品组之前,注入空白溶剂乙腈以确保系统没有污染物或干扰峰。
2.2.2计算方法
样品中的色素含量按照公式(1)计算:
式中:
x:样品中色素含量,单位为克每千克(g/kg);
c:样品中色素的浓度,单位为微克每毫升(μg/ml);
v:样品稀释体积,单位为毫升(ml);
m:样品质量,单位为克(g);
1000:换算系数。
经检测,青梅酒和百香果酒中含有21种色素中的某些成分。具体如图27所示为青梅酒样品的hplc检测图谱。
如图28为百香果酒样品的hplc检测图谱。
其中青梅酒检出色素的出峰时间为:柠檬黄11.8min,亮蓝22.926min。经计算,柠檬黄含量为10.17mg/kg,亮蓝含量为2.24mg/kg,均未超标。(国家标准gb2760-2014规定配制酒中柠檬黄限量0.1g/kg;亮蓝限量0.025g/kg)
含有色素柠檬黄的百香果样品:
百香果酒检出色素的出峰时间为:柠檬黄11.8min。经计算,柠檬黄含量为6.72mg/kg,未超标。
方法可靠性验证
以目标物的色谱保留时间进行定性,以色谱峰的峰面积用标准曲线外标法进行定量。在优化的色谱条件下,21种人工合成色素在0.4μg/ml-20.0μg/ml质量浓度范围内呈现良好线性关系,r2≥0.9997。分析低水平添加溶液的信噪比,计算21种色素的检出限(s/n=3)与定量限(s/n=10),检出限为0.03-0.16mg/kg,定量限为0.10-0.50mg/kg,低、中、高三个不同浓度加标回收率分别为88.1%-106.5%、87.2%-99.6%和89.5%-99.2%,相对标准偏差(rsd%,n=6)分别为1.1%-3.2%、1.2%-3.1%和1.0%-2.9%。
其中21种合成色素在青梅酒和百香果酒样品的回收率数据如下表2。
表221种合成色素在青梅酒和百香果酒样品的低中高三个加标质量浓度分别为0.5mg/kg,2.0mg/kg,10mg/kg的回收率(%)和精密度rsd(%)(n=6)
从表2数据可以看出,本发明提供的方法回收率高,稳定性好,rsd小于3.2%,精确度高。
实施例3
本实施例与实施例1相比,区别仅在于:
(1)聚苯乙烯微球(ps)的制备方法:
100ml三口烧瓶中加入15ml6%苯乙烯单体和30ml水,室温超声分散20min,一次性加入浓度为0.5mol/l的kps水溶液。通氮保护下恒温90℃聚合反应12h,水洗离心烘干。合成ps球粒径大小在200nm左右。pani@ps粒径210±10nm。
采用本发明提供的吸附包对青梅酒和百香果酒中各色素的回收率及精密度进行测定,结果为:低、中、高三个不同浓度加标回收率分别为76.3%-93.1%、89.6%-94.1%和89.3%-95.2%,相对标准偏差(rsd%,n=6)分别为2.1%-5.5%、2.6%-6.1%和2.3%-5.7%。如下表3所示。
表321种合成色素在合成萃取剂大粒径pani@ps前处理条件下的青梅酒和百香果酒样品的低中高三个加标质量浓度分别为0.5mg/kg,2.0mg/kg,10mg/kg的回收率(%)和精密度rsd(%)(n=6)
分析:关于ps粒径,增加反应时间,可以增加粒径半径到200-500nm。
球形颗粒粒径越小,比表面积越大,比表面积随粒径减少急剧增加,小粒径的ps球接触到的活性位点更多,更有利于吸附解析反应,更有利于实验进行。
对比例
如下对比例考察不同前处理工艺对检测结果的影响,与实施例2中样品前处理1)-5)相比,各对比例存在如下区别:
对比例1:原液浓缩离心后直接过0.45μm滤膜上机。
对比例2:聚酰胺吸附法;方法:步骤(1)(2)相同,接着取1.0g聚酰胺固体(颗粒度80-100目)调成粘稠状,倒入样品搅拌,漏斗边抽滤边用水(ph4)和甲醇-甲酸(体积比为6:4)混合溶液循环洗涤数次,直至中性;再用乙醇-氨水-水洗涤数次,直至色素解析完全,解吸液用乙酸中和,蒸发近干,加水复溶,定容过膜。
对比例3:hlb柱固相萃取法;步骤(1)(2)相同,接着上述溶液自然流入hlb柱(watershlb柱,6cc,200mg,partno:wat106202,提前分别用5ml甲醇和0.1%甲酸-水预洗),用6ml浓度为2.0%氨水与甲醇的混合液(二者体积比为3:7)洗脱,收集洗脱液,甲酸中和,氮吹浓缩,加水复溶,定容过膜。
对比例4:聚酰胺柱固相萃取法;步骤(1)(2)相同,接着上述溶液自然流入聚酰胺柱(spe柱,1g,6ml,cnw,sbeq-ca7355),提前分别用5ml甲醇和0.1%甲酸预洗),用6ml甲醇-氨水-水(三者体积比为7:2:1)洗脱,收集洗脱液,甲酸中和,氮吹浓缩,加水复溶,定容过膜。
对比例5:pani@ps直接萃取法;省略步骤(3),仅使用萃取包2进行快速分散萃取。
检测结果如图27、28所示。含有目标物组分的某2种实际样品(青梅酒和百香果酒)在不同前处理条件下(参考对比例1-5)的色谱图,横坐标x为目标物组分浓度,纵坐标y为峰面积响应值。
6种不同前处理条件:1.直接浓缩过膜上机;2.聚酰胺吸附法;3.hlb柱固相萃取法;4.聚酰胺柱固相萃取法;5.pani@ps萃取法;6.本法萃取包
检测结果
方法1-5不同前处理条件下的青梅酒和百香果酒样品的回收率(%)和精密度rsd(%)范围分别为:方法1,回收率70.8%-82.1%,精密度3.4%-9.7%;方法2,回收率75.4%-85.6%,精密度2.5%-7.9%;方法3,回收率79.2%-86.9%,精密度3.3%-6.8%;方法4,回收率80.7%-88.4%,精密度2.9%-8.1;方法5,回收率85.1%-93.5%,精密度2.7%-6.5%。
具体检测结果数据如下表4-1和4-2所示:
表4-121种合成色素在1-5不同前处理条件下的青梅酒样品的回收率(%)(加标质量浓度为2.0mg/kg)和精密度rsd(%)(n=6)
表4-221种合成色素在1-5不同前处理条件下的百香果酒样品的回收率(%)(加标质量浓度为2.0mg/kg)和精密度rsd(%)(n=6)
最后应说明的是:以上仅为本发明的优选实施例而已,并不用于限制本发明,尽管参照前述实施例对本发明进行了详细的说明,对于本领域的技术人员来说,其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!