一种食品中水溶性阴离子合成色素的检测方法与流程
本发明涉及一种快速筛查合成色素的方法,具体地涉及一种食品中水溶性阴离子合成色素检测方法。
背景技术:
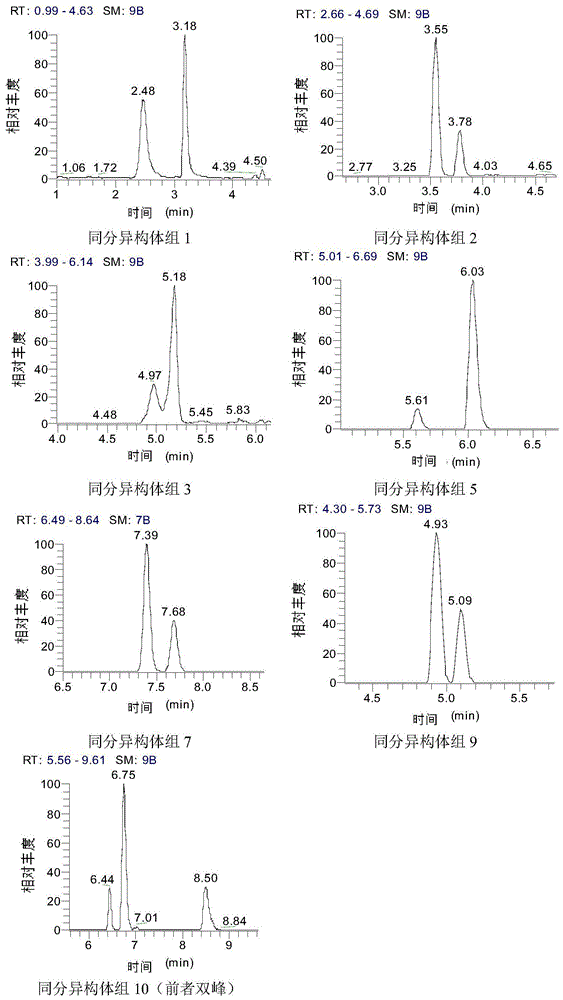
:合成色素主要是以煤焦油中分离出来的苯胺染料为原料制成的着色剂。相比天然色素,合成色素优点不少,如色泽鲜艳、着色力强、色调多样、稳定性更高、保质期长、价格低廉等,目前世界各国允许使用的合成色素几乎都是水溶性色素。酸性合成色素是一类水溶性阴离子合成色素。合成色素分子中含磺酸基、羧基等酸性基团,通常以钠盐的形式存在,在酸性染浴中可以与蛋白质分子中的氨基以离子键结合,故称为酸性合成色素。由于在水中溶解度较大,更容易与食品中的蛋白质及其它成分更好地结合,酸性合成色素如日落黄、诱惑红等在食品工业中的应用更加广泛,适用于饮料、酒类、调味品、肉制品、豆制品、面制品等各类液态和固态食品,容易因操作不当引起超标。并且,酸性合成色素的同分异构体种类繁多,还存在着大量禁止在食品中使用的酸性合成色素同分异构体,如国家允许使用的食品着色剂日落黄、亮蓝就分别存在酸性橙10、酸性绿5两种食品中禁用的同分异构体酸性合成色素,因此更容易在食品中误用或非法添加该类色素。国内外对水溶性合成色素的检测方法研究也多种多样,也在不断改进。其中,超高效液相色谱质谱联用法准确度高,是近年来应用较多的方法。但存在前处理复杂、基质效应大、针对不同检测对象,需要分别调整基质,才能够进行准确的检测、检测种类不多、检测对象较单一等缺点,部分同分异构体色素,因液相保留时间相同,无法有效定性定量,存在的严重问题在于:由于无法区分同分异构体,导致检测员错误地认为部分违禁品添加品是合规的同分异构体从而导致检测结论错误,具有极大的错检漏检风险,这是本领域亟待解决的问题。因此,建立前处理简单、有效降低基质效应、高通量、检测对象广泛、并能准确定性定量检测同分异构体的检测方法成为了食品中合成色素检测迫切需要解决的技术问题。技术实现要素:针对现有技术的上述缺陷,本发明提供了一种能同时对食品中的多种水溶性阴离子合成色素进行检测方法。本发明利用四级杆静电子轨道离子阱质谱分辨率高的优势,结合液相色谱分离手段,根据同分异构体二级质谱的差异,实现了对含有多组同分异构体的92种水溶性阴离子合成色素的定性和定量,可以实现食品中允许使用与禁止使用的水溶性合成色素同分异构体的鉴定。本发明的食品中水溶性阴离子合成色素的检测方法,所述方法包括以下步骤:1)、提取样品,净化、稀释得测试液;2)、用液相色谱-四级杆静电子轨道离子阱质谱对步骤1)所得测试液进行测定,以标准溶液建立校正曲线,外标法定量。进一步地,本发明所述水溶性阴离子合成色素的名称及定量离子为:柠檬黄,232.99501;新红,271.48616;苋菜红,267.98325;靛蓝,209.98665;胭脂红,267.98325;喹啉黄,215.48903;日落黄,202.99702;诱惑红,225.01013;亮蓝,373.07187;偶氮玉红,228.00484;赤藓红,834.64777;色酚黄s,155.98497;酸性红1,231.50775;酸性红73,511.03876;刚果红,325.05266;酸性蓝1,543.16290;橙黄ii,327.04450—>170.99997;橙黄iv,352.07614;甲基橙,304.07614;直接黄8,507.08023;酸性黄17,251.96895;酸性红52,557.14217;酸性红9,377.06015;酸性红13,228.00484—>221.01559;酸性红87,646.69915;酸性红88,377.06015—>143.0504;酸性红91,578.85033;酸性红92,784.54031;酸性红94,972.48893;酸性红151,431.08195;酸性红265,294.51472;酸性红289,326.06744;铬变素2b,233.48702;铬变素2r,210.99448;酸性红26,435.03262;酸性橙6,293.02377;酸性橙20,327.04450;藏花橙g,327.04450—>206.99979;酸性橙17,355.07580;二甲苯青ff515.1316;酸性蓝83,400.62767;酸性蓝90,414.64332;酸性蓝3,559.15782;酸性蓝92,313.50435;酸性绿16,593.17855;酸性绿5,373.07187;酸性绿9,701.15523;酸性绿27,330.08055;酸性绿50,553.11087;酸性绿a,571.19420;酸性绿25,288.03360;酸性黑1,285.01373;酸性紫3,438.00713;酸性紫34,577.07448;酸性紫49,710.23640;酸性黄36,352.07614;酸性橙10,407.00132;酸性红44,457.01697—>301.95639;酸性红17,228.00484;棕色ht,303.02631;胭脂红sx,435.03262;酸性紫9,589.14388;坚牢绿,381.06933;食品黑1,258.65832;刚果红d8,329.07777;酸性橙iid6,333.08216;丽春红3r,224.02049;滂酰紫6r,236.00230;荧光素钠,331.06120;滂酰洋红2b,246.51303;酸性红60,275.98071;酸性棕14,288.02103;酸性蓝113,317.54721;酸性红66,255.01574;酸性绿41,304.02852;酸性黄11,357.06630;酸性橙8,341.06015;酸性红71,269.03139;酸性黄9—钠盐,177.49719;酸性蓝93,376.54602;酸性蓝7,667.19420;颜料红49,377.06015—>297.1037;酸性蓝62,399.10202;酸性紫7,260.01848;酸性红50,529.11087;酸性红33,210.50247;酸性蓝41,464.09218;酸性黑210,429.54540;酸性兰27,421.08637;溶剂蓝37,313.50435;乙基曙红,674.73045;亮黄,289.02885,来自于表4。进一步地,本发明所述提取包括以下步骤:称取固体样品1~2g,加入5~10ml甲醇,涡旋,离心,回收上清液,加入5~10ml提取液重复提取一次,合并上清液,用甲醇定容至25ml;准确移取2ml定容后提取液并氮吹至干,用甲醇复溶,得样品提取液;优选地,所述提取液为氨水和甲醇的混合液,所述混合液中氨水和甲醇体积比(1~20):(80~99)。或者,本发明所述提取包括以下步骤:准确移取液体样品1~2ml,用甲醇稀释12~13倍,得样品提取液。进一步地,本发明所述净化步骤为:移取1ml样品提取液于分散固相萃取小管净化,涡旋,离心;优选地,所述分散固相萃取小管为c18萃取小管。进一步地,本发明所述稀释包括以下步骤:取离心所得上清液用水进行稀释,获得测试液;优选地,所述上清液与水的稀释比为1:(8~10)。进一步地,本发明所述液相色谱-四级杆静电子轨道离子阱质谱条件为:高效液相色谱条件:色谱柱:thermofisheraccucoreaqc18,150mm×2.1mm,2.6μm;保护柱accucoreaq10×2.1mm,2.6μm;流动相a为5mm乙酸铵(含5%甲醇),流动相b为甲醇;梯度洗脱;柱温:40℃;进样量:1μl;流速:0.3ml/min;质谱条件:离子源参数:负离子模式,sprayvoltage(-)喷雾电压3.0kv,capillarytemperature离子传输管温度320℃,sheathgas鞘气流速40arb,auxgas辅助气流速10arb,probeheatertemperature,喷雾针温度350℃,s-lensrflevel离子透镜电压55;fullms(一级质谱全扫描)模式:质谱分辨率为70000,agctarget(自动增益控制目标)为3e6,maximumit(最大驻留时间)为200ms,扫描范围100-1000m/z。进一步地,本发明所述质谱条件包括平行反应监测模式,其条件为:质谱分辨率为17500,agctarget(自动增益控制)为2e5,maximumit(最大驻留时间)为100ms,isolationwindow(隔离窗口)为1.0m/z,扫描范围50-母离子*电荷数+20.进一步地,本发明以体积百分比计,所述梯度洗脱条件为:从0~0.5min,a相为100%;从0.5~4.0min,a相由100%均匀变化至55%;从4.0~8.0min,a相为55%;从8.0~13.0min,a相由55%均匀变化至25%;从13.0~14.0min,a相由25%均匀变化至2%;从14.0~17.0min,a相为2%;从17.0~17.1min,a相由2%均匀变化至100%;从17.1~20.0min,a相为100%。进一步地,本发明质谱分析时,当化合物的最高响应离子不稳定或存在杂质干扰时,选择稳定性高且无干扰的离子作为其定量离子。本发明相对于现有技术具有如下有益效果:(1)、本发明应用范围较广:检测食品包括各类液态和固态食品,一次进样可同时检测的水溶性合成色素种类共92种,包含10组24种同分异构体。(2)、本发明能同时对多组同分异构体水溶性合成色素进行色谱分离、准确定性和定量,解决了食品中同分异构体色素难以区分、无法同时检测定量的检测难题。同时能有效提高对违禁添加色素的检测准确性,确保违法添加物质与合法添加物质存在同分异构体时由于无法区分导致的检测结论误差,保证检测的质量和准确性。(3)、本发明所的样品前处理方法利用分散固相萃取净化方法,快速去除样品基质中脂肪等杂质,同时结合大比例稀释的方法降低样品基质效应,避免了使用内标或基质标准。(4)、本发明采用标准溶液外标法定量,不依赖同位素内标(不易购买,成本高昂)或基质曲线标准(配制繁琐,不同食品种类需配制不同基质曲线)。(5)、本发明利用水溶性阴离子合成色素具有多种离子形态(不同价态)一级母离子的特点,灵活调整化合物的定量离子,当化合物其中一种形态母离子受到杂质干扰时,通过选择另一种形态、无杂质干扰的母离子进行定量,从而避免杂质带来的影响。附图说明图1液相条件下保留时间不同的七组同分异构体色谱图。图2液相条件下保留时间重叠的三组同分异构体色谱图。图3三组保留时间相同的同分异构体的二级质谱图及特征碎片离子色谱图。图4溶剂蓝3不同母离子定量的色谱峰和定量校正曲线。图5果酒基质中酸性红26两个母离子的质谱采集点数与其附近干扰离子。图6六种典型样品不同稀释比下基质效应。具体实施方式为了更好地说明本发明的技术目的、技术方案和优点,现结合附图与具体实施例对本发明做进一步说明。一种食品中水溶性阴离子合成色素(包括92种合成色素,具体见下表1)检测方法,所述方法包括如下步骤:表1水溶性阴离子合成色素名称、cas号和分子式(1)、配制标准工作溶液上述92种合成色素根据其化学性质用甲醇、乙腈等有机溶剂配制标准储备溶液,再用甲醇-水(1:9)配制标准工作溶液。(2)、样品前处理a)提取固体:准确称取2g固体样品,加入10ml甲醇,涡旋,离心,回收上清液,加入10ml氨水和甲醇的混合液(氨水和甲醇的混合体积比(1~20):(80~99))重复提取一次,合并提取液,用甲醇定容,准确移取2ml定容后的提取液于氮吹管中氮吹干,用甲醇复溶,获得样品提取液。液体:准确移取2ml液体样品,用甲醇稀释12~13倍,得样品提取液。b)净化移取1ml样品提取液于分散固相c18萃取小管净化,涡旋,离心。c)稀释取离心所得上清液用水进行稀释,获得测试液;优选地,所述上清液与水的稀释比为1:(8~10)。(3)、用液相色谱-四级杆静电子轨道离子阱质谱对步骤(2)所得测试液进行测定,以标准溶液建立校正曲线,外标法定量。采用超高效液相色谱四级杆静电子轨道离子阱质谱测定,根据峰面积和浓度建立校正曲线。其中,超高效液相色谱-四级杆静电子轨道离子阱质谱检测条件如下:质谱条件为:离子源参数:负离子模式,sprayvoltage(-)喷雾电压3.0kv,capillarytemperature离子传输管温度320℃,sheathgas鞘气流速40arb,auxgas辅助气流速10arb,probeheatertemperature,喷雾针温度350℃,s-lensrflevel离子透镜电压55。采集模式和参数:采用fullms(一级质谱全扫描)+prm(平行反应监测)模式,fullms(一级质谱全扫描)质谱分辨率为70000,agctarget(自动增益控制目标)为3e6,maximumit(最大驻留时间)为200ms,扫描范围100-1000m/z;prm(平行反应监测)质谱分辨率为17500,agctarget(自动增益控制)为2e5,maximumit(最大驻留时间)为100ms,isolationwindow(隔离窗口)为1.0m/z,扫描范围50-母离子*电荷数+20。在上述的质谱采集模式中,fullms是对一级质谱进行全扫,prm是针对特定离子做预设碰撞能量(具体请见表3)下的二级质谱全扫。本发明正是通过以上两种质谱采集模式,实现灵活运用两种模式定量,一种是采用一级母离子定量,适用于非同分异构体的合成色素和保留时间不同的同分异构体色素;另一种是采用二级碎片离子定量,适用于保留时间相同的同分异构体色素,即在对待检测样品进行一级质谱进行全扫的过程后,还采用prm平行反应监测模式进行检测,其达到的技术效果是可以避免一级质谱进行全扫中可能存在的干扰从而利用prm平行反应监测模式进行检测进行检测,确保了检测的准确性。10组同分异构体的分离本发明所述的92种水溶性阴离子合成色素中共包含了10组24种互为同分异构体色素,具体见下表2。表2同分异构体色素序号、名称、分子式和结构式通过本申请的检测方式,采用fullms(一级质谱全扫描)+prm(平行反应监测)模式得到检测结论参见表2。由表2可知,这些分子式完全相同的同分异构体合成色素中既有国家允许在食品中使用的合成色素,也有国家禁止在食品添加的合成色素,比如,组别2中的日落黄、组别3中的亮蓝、组别4中的偶氮玉红,他们均是允许使用的色素,而他们分别对应的同分异构体色素——酸性橙10、酸性绿5、酸性红13、酸性红44和酸性红17则是禁用色素。而现有的国家检测标准《食品安全国家标准食品中合成着色剂的测定(gb5009.35-2016)》仅针对允许使用的合成色素检测,无法对与他们互为同分异构体的禁用色素进行区分和检测,造成监管部门不能及时发现可能存在的添加禁用色素的违法行为,导致食品安全监管的漏洞。因此,为了解决上述现有技术存的检测标准的不足,本发明通过下述液相色谱洗脱条件和2种定量模式的方法对上述10组同分异构体色素进行分离和准确定量。首先通过选用特定的液相色谱洗脱条件,实现保留时间上的分离,并采用一级母离子进行定量;再对液相无法分离、保留时间相同的同分异构体色素使用prm模式采集,通过提取各化合物独有的二级碎片离子进行分别定量。本发明特定的液相色谱条件如下:色谱柱:thermofisheraccucoreaqc18(150mm×2.1mm,2.6μm)+保护柱accucoreaq(10×2.1mm,2.6μm);流动相a为5mm乙酸铵(含5%甲醇),流动相b为甲醇;梯度洗脱;柱温:40℃;进样量:1μl;流速:0.3ml/min;梯度洗脱:从0~0.5min,a相为100%;从0.5~4.0min,a相由100%均匀变化至55%;从4.0~8.0min,a相为55%;从8.0~13.0min,a相由55%均匀变化至25%;从13.0~14.0min,a相由25%均匀变化至2%;从14.0~17.0min,a相为2%;从17.0~17.1min,a相由2%均匀变化至100%;从17.1~20.0min,a相为100%。在上述的液相色谱条件下,10组同分异构体中,共有7组(同分异构体组1、2、3、5、7、9、10)实现了色谱分保留时间的分离(见图1),由图1可知,允许使用的色素日落黄与禁用色素酸性橙10,保留时间分别为3.78和3.55,允许使用的色素亮蓝与禁用色素酸性绿5,保留时间分别为5.18和4.97,允许使用的色素偶氮玉红与禁用色素酸性红13、酸性红44和酸性红17,保留时间分别为5.04、4.86(酸性红13、酸性红44重叠)和5.17,允许使用的色素与禁用色素均实现了色谱分离,实现了禁用色素与非禁用色素的同时检测。剩余3组保留时间相近或几乎重叠(见图2),采用prm模式利用二级碎片定量。为更好说明prm定量模式,以同分异构体橙黄ii(酸性橙ii)和藏花橙g为例,该二者母离子均为327.04450,保留时间均为6.8min,但是二者分别具有对方所不能产生的特征二级碎片离子170.99997(橙黄ii独有)和206.99979(藏花橙g独有),通过提取以上两个碎片离子实现橙黄ii和藏花橙g的区分和分别定量。表3为针对该3组同分异构体的prm中inclusionlist参数。图3是3组保留时间相同的同分异构体二级质谱图及特征碎片离子色谱图。表3prm(平行反应监测)中inclusionlist设置参数化合物母离子(m/z)开始时间(min)结束时间(min)碰撞能量ce酸性橙ii&藏花橙g327.044504.508.0025酸性红88&颜料红49377.060159.5013.0028酸性红44457.016973.506.5020酸性红13228.004843.506.5015定量离子的优化本发明在选择各化合物定量离子时,优选化合物的最高响应离子,当最高响应离子不稳定或存在杂质干扰时,则选择稳定性高且无干扰的离子作为其定量离子。如本实施例中,在建立校正曲线时,发现溶剂蓝37采用最高响应的三价母离子208.66743(离子形态[m-3na]3-)定量时,峰形和峰面积的稳定性差,校正曲线相关系数仅有0.963,采用次级响应的二价母离子313.50486(离子形态[m-3na+h]2-),峰形和峰面积的稳定性均良好,校正曲线相关系数能达到0.999,具体见图4是溶剂蓝37不同母离子定量的色谱峰和定量校正曲线。即通过选择不同母离子定量,有利于保证检测结论的准确性。基于相同的基理,申请人发现在检测果酒的过程中,酸性红26如果采用响应最高的一级母离子定量217.01256(离子形式为[m-2na]2-),色谱峰采集点数不足导致峰响应重复性差,通过查看一级全扫质谱图分析发现,在217.01256附近存在一个响应很强的干扰离子217.00336,而酸性红26响应次高的一级母离子435.03262(离子形式为[m-2na+h]-)附近则没有干扰离子的影响,扫描点数大于12(具体见图5)。因此,本发明在建立酸性红26校正曲线时,选择435.03262作为定量离子,避免了样品中杂质的干扰。故本发明最终确定92种合成色素定量离子的具体参数见下表4。基质效应考察分别选取葡萄酒、果酒、番茄酱、腐竹、肉脯和辣椒面六种典型样品进行检测,分别称取2g固体样品(液体样品移取2ml)加入10ml甲醇提取(液体用甲醇定容至10ml),获得六种样品空白基质提取液(即样品稀释倍数为5倍的空白基质液),然后用甲醇-水(1:9)对空白基质提取液进行10倍和20倍稀释(即样品稀释倍数为50倍和100倍的空白基质液),再分别用不同稀释倍数的空白基质液配制基质标准,同时采用甲醇-水(1:9)配制溶剂标准,通过比较不同稀释倍数基质标准和溶剂标准的斜率,研究不同稀释倍数下(5倍、50倍、100倍)样品基质效应的变化规律,确定能够消除样品基质效应的最优稀释倍数(见图6)。实验结果表明,当样品的稀释比为1:100时,92种合成色素的基质效应基本均降低至±20%的可接受水平范围。(4)线性范围、校正方程、相关系数、检出限实施例1本申请检测方法针对腐竹和果酒合成色素加标回收试验:1)样品加标a)样品提取:腐竹:准确称取2.00g腐竹样品于50ml离心管中,向样品中加入适量体积合成色素标准溶液(使最终测试液的浓度分别为线性范围1倍、2倍和10倍最低浓度点con.min),加入10ml甲醇,涡旋,在转速8000r/min下离心3min,转移上清液于25ml比色管中,加入10ml5%氨水-甲醇重复提取一次,合并提取液,用甲醇定容,准确移取2ml定容后提取液到氮吹管中,氮吹至近干,用2ml甲醇复溶,获得样品提取液。果酒:准确移取2ml样品于25ml比色管中,用甲醇定容,获得样品提取液。b)净化:移取1ml样品提取液于分散固相c18萃取小管净化,涡旋,离心。c)稀释:取上清液用蒸馏水进行稀释,稀释比为1:8,获得测试液。2)标准溶液配制及样品测定吸取92种色素混合标准溶液,用甲醇-水(1:9)配制标准工作溶液,标准工作液和样品测试液采用超高效液相色谱四级杆静电子轨道离子阱质谱测定,根据峰面积和浓度建立校正曲线,对样品进行定量分析。质谱条件:离子源参数:负离子模式,sprayvoltage(-)喷雾电压3.0kv,capillarytemperature离子传输管温度320℃,sheathgas鞘气流速40arb,auxgas辅助气流速10arb,probeheatertemperature,喷雾针温度350℃,s-lensrflevel离子透镜电压55;fullms(一级质谱全扫描)模式:质谱分辨率为70000,agctarget(自动增益控制目标)为3e6,maximumit(最大驻留时间)为200ms,扫描范围100-1000m/z。prm(平行反应监测)模式:质谱分辨率为17500,agctarget(自动增益控制)为2e5,maximumit(最大驻留时间)为100ms,isolationwindow(隔离窗口)为1.0m/z,扫描范围50-母离子*电荷数+20。高效液相色谱条件:色谱柱:thermofisheraccucoreaqc18(150mm×2.1mm,2.6μm);保护柱accucoreaq(10×2.1mm,2.6μm);流动相a为5mm乙酸铵(含5%甲醇),流动相b为甲醇;梯度洗脱;柱温:40℃;进样量:1μl;流速:0.3ml/min;洗脱条件为:从0~0.5min,a相为100%;从0.5~4.0min,a相由100%均匀变化至55%;从4.0~8.0min,a相为55%;从8.0~13.0min,a相由55%均匀变化至25%;从13.0~14.0min,a相由25%均匀变化至2%;从14.0~17.0min,a相为2%;从17.0~17.1min,a相由2%均匀变化至100%;从17.1~20.0min,a相为100%。结果分析腐竹和果酒样品加标回收试验结果见表5。实施例1证明:本发明的方法测定92中水溶性阴离子合成色素在线性范围内具有良好的线性关系,相关系数均大于0.99,方法检出限在0.01-0.4μg/kg之间。实际样品中高中低三个添加水平下,平均回收率(recovery)基本在60%-110%范围内,相对标准偏差(rsd)为0.1%-24.47%,小于25%,具有较好的方法灵敏度、准确度和精密度。即通过加标回收验证了本申请检测方案结论的准确性。实施例2对市售调味品和饮料酒样品中92种水溶性阴离子合成色素的筛查。1)样品处理a)样品提取:固体样品:准确称取2.00g于50ml离心管中,加入10ml甲醇,涡旋,在转速8000r/min下离心3min,转移上清液于25ml比色管中,加入10ml5%氨水-甲醇重复提取一次,合并提取液,用甲醇定容,准确移取2ml提取液到氮吹管中,氮吹至近干,用2ml甲醇复溶,获得样品提取液。液体样品:准确移取2.00ml样品于25ml比色管中,用甲醇定容,获得样品提取液。b)净化:移取1.00ml样品提取液于分散固相萃取小管净化,涡旋,离心。其中,分散固相萃取小管为c18萃取小管。c)稀释:取上清液用蒸馏水进行稀释,获得测试液。稀释比为1:8。2)标准溶液配制及样品测定同实施例1。3)结果分析本实施例对市售的50个调味品样品和102个饮料酒样品中92种水溶性阴离子合成色素进行筛查,共有2个调味品和1个饮料酒检出了含有水溶性阴离子合成色素,其中调味品检出的均为国家允许使用的食品着色剂,分别是日落黄及诱惑红。饮料酒中检出了苋菜红、亮蓝和酸性红13,其中苋菜红和亮蓝为允许使用的色素,酸性红13为禁用色素,其与国家允许使用的色素偶氮玉互为同分异构体,在本发明所述的方法下,准确地将该二者区分并定量。饮料酒中酸性红13的发现,也说明了市场确实存在违法使用禁用色素替代允许使用色素的可能,阳性样品筛查结果具体见表7。表7阳性样品检测结果以上实施例虽然未能包含本发明在各种食品中的应用,但通过本发明的思路,可有效去除基质效应的影响,检测各种不同类型和基质的待处理样品,并且由于能够分辨出同分异构体,确保了检测结论的准确性,具有极大的推广意义。本发明样品前处理操作简便、快速,无需使用基质标准或内标物定量。本发明检测方法采用两种定量模式,先采用一级母离子定量,检测将非同分异构体的合成色素和保留时间不同的同分异构体色素;再采用二级碎片离子定量,对保留时间相同的同分异构体色素进行准确的定性定量检测,填补了现有的检测方法无法同时测定食品中多种合成色素及其同分异构体的缺失。上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制。其它的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含本发明的保护范围之内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1