一种石斛夜光丸的含量测定方法与流程
本发明涉及中医药质量检测技术领域,具体是一种石斛夜光丸的含量测定方法。
背景技术:
石斛夜光丸由石斛、人参、山药、茯苓、甘草、肉苁蓉、枸杞子、菟丝子、地黄、熟地黄、五味子、天冬、麦冬、苦杏仁、防风、川芎、麸炒枳壳、黄连、牛膝、菊花、盐蒺藜、青葙子、决明子、水牛角浓缩粉、山羊角等二十五味中药组成。除水牛角浓缩粉外,山羊角锉研成细粉,其余石斛等二十三味药材粉碎成细粉;将水牛角浓缩粉与上述粉末配研,过筛,混匀。每100g粉末用炼蜜35~50g加适量的水制丸,干燥,制成水蜜丸;或加炼蜜95~120g制成小蜜丸或大蜜丸,即得。石斛夜光丸具滋阴补肾、清肝明目之功效;临床用于肝肾两亏,阴虚火旺,内障目暗,视物昏花。石斛夜光丸现行标准【含量测定】控制的指标为黄连中的盐酸小檗碱,不能有效、整体性评价石斛夜光丸的质量。
贾元印等人以北京、济南、广州等7个厂家石斛夜光丸为样品,用薄层扫描法测定样品中人参二醇及人参三醇的含量,建立了测定方法,为制定石斛夜光丸的质控标准提供了参考(贾元印,赵渤年,刘青,刘永俊.石斛夜光丸中人参二醇及人参三醇的含量测定.中国中药杂志,1995,20(12):732-735.)。但是由于薄层扫描法容易受温度、薄层板、点样量等因素影响,含量测定结果准确率不稳定,且人参的主要有效成分为人参皂苷,而该文章中是以人参皂苷的水解产物人参二醇及人参三醇为指标进行含量测定的,并试验了多种方法均显示由于成分干扰严重并不能直接测定样品中主要皂苷的含量,因此,其方法并不能准确反应石斛夜光丸中人参的真实质量,依然需要进行深入研究寻找到适用于建立石斛夜光丸含量检测标准的方法。
技术实现要素:
本发明提供了一种石斛夜光丸的含量测定方法,本发明通过高效液相色谱法建立柚皮苷、新橙皮苷、人参皂苷rg1、人参皂苷re与人参皂苷rb1的含量测定方法,以考察石斛夜光丸中人参、麸炒枳壳的质量情况,能快速、准确地检查出企业是否存在不投料或少投料、伪劣药材投料的行为。
一种石斛夜光丸的含量测定方法,具体步骤如下:
(1)色谱条件与系统适用性试验:以十八烷基硅烷键合硅胶为填充柱;以乙腈为流动相a,以水为流动相b,按以下表1进行梯度洗脱;流速为1.0ml/min;用高温型蒸发光散射检测器检测,理论板数按人参皂苷rg1峰计算应不低于3000。
(2)混合对照品溶液制备:精密称取柚皮苷、新橙皮苷、人参皂苷rg1、人参皂苷re与人参皂苷rb1适量,加70%甲醇制成每1ml含柚皮苷与新橙皮苷各0.5mg、人参皂苷rg1与人参皂苷re各0.1mg、人参皂苷rb10.2mg的混合对照品溶液。
(3)供试品溶液的制备:取石斛夜光丸水蜜丸,研碎,取6g;或取石斛夜光丸小蜜丸或大蜜丸,剪碎,取9g;精密称定,置具塞锥形瓶中,加入体积浓度为70%的甲醇溶液100ml,称定重量,加热回流1h,取出,放冷,再称定重量,用体积浓度为70%的甲醇溶液补足减失的重量,摇匀,滤过,精密吸取续滤液50ml,蒸干,残渣加水20ml使溶解,用水饱和的正丁醇振摇提取4次,每次25ml,取正丁醇液,蒸干,残渣加甲醇溶解并转移至5ml量瓶中,加甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。
(4)测定:分别精密吸取混合对照品溶液5μl、15μl,供试品溶液10μl,注入液相色谱仪,测定,以外标两点法对数方程计算含量。
以上所述石斛夜光丸中含麸炒枳壳以柚皮苷(c27h32o14)和新橙皮苷(c28h34o15)计,水蜜丸含柚皮苷每1g不得少于0.42mg,含新橙皮苷每1g不得少于0.31mg;小蜜丸含柚皮苷每1g不得少于0.28mg,含新橙皮苷每1g不得少于0.21mg;大蜜丸含柚皮苷每丸不得少于1.57mg,含新橙皮苷每丸不得少于1.18mg;含人参以人参皂苷rg1(c42h72o14)、人参皂苷re(c48h82o18)、人参皂苷rb1(c54h92023)计,水蜜丸含人参皂苷rg1和人参皂苷re的总量每1g不得少于0.13mg,含人参皂苷rb1每1g不得少于0.084mg;小蜜丸含人参皂苷rg1和人参皂苷re的总量每1g不得少于0.086mg,含人参皂苷rb1每1g不得少于0.057mg;大蜜丸含人参皂苷rg1和人参皂苷re的总量每丸不得少于0.47mg,含人参皂苷rb1每丸不得少于0.31mg。
本发明的有益效果:
本发明通过高效液相色谱法建立柚皮苷、新橙皮苷、人参皂苷rg1、人参皂苷re与人参皂苷rb1的含量测定方法,以考察石斛夜光丸中人参、麸炒枳壳的质量情况,能快速、准确地检查出企业是否存在不投料或少投料、伪劣药材投料的行为。而且在本发明的含量测定方法中,采用高温型蒸发光散射检测器进行检测,干扰峰较小,人参皂苷rg1、人参皂苷re与人参皂苷rb1响应值较高,分离度好。通过增加本发明的含量测定方法作为石斛夜光丸现行标准【含量测定】的补充,可以使石斛夜光丸的评价标准更为全面,能有效确保石斛夜光丸用药安全,减少不投料或掺伪投料的现象,为石斛夜光丸的质量标准研究及其质量控制提供科学依据。
附图说明
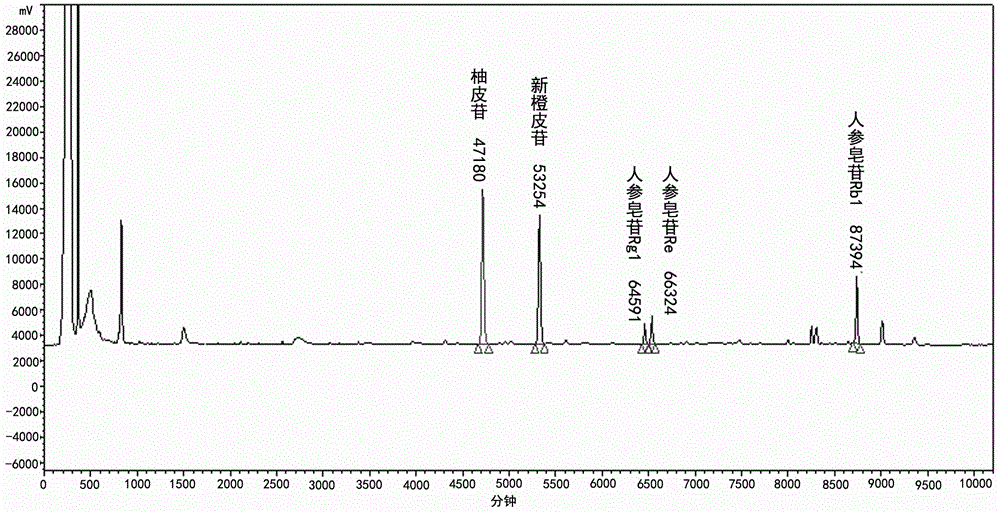
图1是混合对照品溶液的高效液相色谱图。
图2是石斛夜光丸样品(b制药厂,批号18013508)的高效液相色谱图。
图3是石斛夜光丸样品(i公司,批号191004)的高效液相色谱图。
图4是石斛夜光丸样品(n公司,批号191001)的高效液相色谱图。
图5是石斛夜光丸缺人参阴性样品的的高效液相色谱图。
图6是石斛夜光丸缺麸炒枳壳阴性样品的高效液相色谱图。
具体实施方式
为了更加详细的介绍本发明,下面结合实施例,对本发明做进一步说明。
实施例
仪器与试药:
waters高效液相色谱仪,高温型蒸发光散射检测器;ml204型电子分析天平(梅特勒-托利多仪器上海有限公司)。
柚皮苷(中国食品药品检定研究院提供,供含测用,批号:110722-201815,含量91.7%)
新橙皮苷(中国食品药品检定研究院提供,供含测用,批号:11857-201804,含量99.4%)
人参皂苷rg1(中国食品药品检定研究院提供,供含测用,批号:110703-201933,含量93.4%)
人参皂苷re(中国食品药品检定研究院提供,供含测用,批号:110754-202028,含量93.9%)
人参皂苷rb1(中国食品药品检定研究院提供,供含测用,批号:110704-202028,含量93.1%)
乙腈为色谱纯,其他试剂均为分析纯。
91批石斛夜光丸(其中74批为水蜜丸,15批为大蜜丸,2批为小蜜丸)均为2020年国家评价性抽验的样品。
方法考察所用石斛夜光丸(水蜜丸)为r公司,批号:1904030;石斛夜光丸(大蜜丸)为b制药厂,批号:18013507。
一种石斛夜光丸的含量测定方法,具体步骤如下:
(1)色谱条件与系统适用性试验:以十八烷基硅烷键合硅胶为填充柱;以乙腈为流动相a,以水为流动相b,按以下表1进行梯度洗脱;流速为1.0ml/min;用高温型蒸发光散射检测器检测,理论板数按人参皂苷rg1峰计算应不低于3000。
(2)混合对照品溶液制备:精密称取柚皮苷、新橙皮苷、人参皂苷rg1、人参皂苷re与人参皂苷rb1适量,加70%甲醇制成每1ml含柚皮苷与新橙皮苷各0.5mg、人参皂苷rg1与人参皂苷re各0.1mg、人参皂苷rb10.2mg的混合对照品溶液。
(3)供试品溶液的制备:取石斛夜光丸水蜜丸,研碎,取约6g;或取石斛夜光丸小蜜丸或大蜜丸,剪碎,取约9g;精密称定,置具塞锥形瓶中,加入体积浓度为70%的甲醇溶液100ml,称定重量,加热回流1h,取出,放冷,再称定重量,用体积浓度为70%的甲醇溶液补足减失的重量,摇匀,滤过,精密吸取续滤液50ml,蒸干,残渣加水20ml使溶解,用水饱和的正丁醇振摇提取4次,每次25ml,取正丁醇液,蒸干,残渣加甲醇溶解并转移至5ml量瓶中,加甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。
(4)测定:分别精密吸取混合对照品溶液5μl、15μl,供试品溶液10μl,注入液相色谱仪,测定,以外标两点法对数方程计算含量。
本申请人采用本发明的方法对对18家生产企业、去重批号后的91批石斛夜光丸进行含量测定,测定结果见表2~4,混合对照品溶液的高效液相色谱图见图1,其中3个生产企业样品典型高效液相色谱图见图2~4。
检测结果显示,13批样品中柚皮苷含量不符合规定,不合格率85.7%;15批样品中新橙皮苷含量不符合规定,不合格率83.5%;1批样品中人参皂苷rg1和人参皂苷re的总量含量不符合规定,不合格率98.9%;1批样品人参皂苷rb1含量不符合规定,不合格率98.9%。其中i公司、k公司、h公司3家生产企业生产的样品中柚皮苷、新橙皮苷未检出,提示该3家生产企业在石斛夜光丸的生产中麸炒枳壳的投料存在问题;d公司、g公司、h公司、l公司、n公司、i公司、j公司、q公司8家生产企业的部分批次人参皂苷rb1相对含量偏高,提示在石斛夜光丸的生产中人参的投料存在问题。
使用spss数据分析软件对各生产企业含量测定结果进行统计分析,结果发现,b制药厂各组分含量测定结果,p值均大于0.05,说明各批次之间含量测定结果均无显著性差异,企业对原药材质量控制较严格,生产工艺较稳定。其他各生产企业的石斛夜光丸柚皮苷、新橙皮苷、人参皂苷rg1、人参皂苷re与人参皂苷rb1含量测定结果,p值均小于0.05,表明各生产企业之间5种成分均有显著性差异,企业对原药材质量控制不严及生产工艺欠稳定。
另外,本申请人还对本发明的含量测定方法进行了专属性和方法学考察,具体如下:
一、专属性考察
按石斛夜光丸处方比例制备一定量的缺人参阴性样品,研碎,取约6g,精密称定,置具塞锥形瓶中,加入体积浓度为70%的甲醇溶液100ml,称定重量,加热回流1h,取出,放冷,再称定重量,用体积浓度为70%的甲醇溶液补足减失的重量,摇匀,滤过,精密吸取续滤液50ml,蒸干,残渣加水20ml使溶解,用水饱和的正丁醇振摇提取4次,每次25ml,取正丁醇液,蒸干,残渣加甲醇溶解并转移至5ml量瓶中,加甲醇稀释至刻度,摇匀,滤过,取续滤液,即得缺人参阴性样品溶液。吸取缺人参阴性样品溶液10μl,注入液相色谱仪,测定,分析,结果缺人参阴性样品在人参皂苷rg1、人参皂苷re、人参皂苷rb13种对照品成分色谱峰相应位置无对应色谱峰,表明阴性无干扰,本发明的方法专属性好,见图5。
按石斛夜光丸处方比例称取一定量的缺麸炒枳壳阴性样品,研碎,取约6g,精密称定,置具塞锥形瓶中,加入体积浓度为70%的甲醇溶液100ml,称定重量,加热回流1h,取出,放冷,再称定重量,用体积浓度为70%的甲醇溶液补足减失的重量,摇匀,滤过,精密吸取续滤液50ml,蒸干,残渣加水20ml使溶解,用水饱和的正丁醇振摇提取4次,每次25ml,取正丁醇液,蒸干,残渣加甲醇溶解并转移至5ml量瓶中,加甲醇稀释至刻度,摇匀,滤过,取续滤液,即得缺麸炒枳壳阴性样品溶液。吸取缺麸炒枳壳阴性样品溶液10μl,注入液相色谱仪,测定,分析,结果缺麸炒枳壳阴性样品在柚皮苷、新橙皮苷2种对照品成分色谱峰相应位置无对应色谱峰,表明阴性无干扰,本发明的方法专属性好,见图6。
二、方法学考察
1、线性关系的考察
(1)精密称取人参皂苷rg1对照品10.73mg,置10ml量瓶中,加甲醇稀释至刻度,摇匀,即得人参皂苷rg1储备液(浓度为1.0022mg/ml)。
(2)精密称取人参皂苷re对照品9.82mg,置10ml量瓶中,加甲醇稀释至刻度,摇匀,即得人参皂苷re储备液(浓度为0.9221mg/ml)。
(3)精密称取人参皂苷rb1对照品10.93mg,置5ml量瓶中,加甲醇稀释至刻度,摇匀,即得人参皂苷rb1储备液(浓度为2.0352mg/ml)。
(4)精密称取柚皮苷对照品9.99mg、新橙皮苷对照品11.53mg,置同一20ml量瓶中,精密加入人参皂苷rg1、人参皂苷re与人参皂苷rb1对照品储备液各2.00ml,加甲醇稀释至刻度,摇匀,即得对照品混合溶液(柚皮苷对照品0.4580mg/ml,新橙皮苷对照品0.5730mg/ml,人参皂苷对照品rg10.1002mg/ml、人参皂苷对照品re0.09221mg/ml、人参皂苷对照品rb10.2035mg/ml)。
(5)分别精密吸取对照品混合溶液2μl、5μl、10μl、15μl、20μl,注入液相色谱仪,测定,以峰面积对数值为纵坐标,对照品进样量对数值为横坐标,绘制标准曲线。
结果见表5,结果表明:柚皮苷对照品在0.9161g~9.161μg,新橙皮苷对照品在1.146g~11.46μg,人参皂苷rg1对照品在0.2004g~2.004μg,人参皂苷re对照品在0.1844g~1.844μg,人参皂苷rb1对照品在0.4070g~4.070μg,范围内线性关系良好。
2、重复性考察
取石斛夜光丸(水蜜丸)适量,研碎,取约6g,精密称定,共取6份,分别置具塞锥形瓶中,加入体积浓度为70%的甲醇溶液100ml,称定重量,加热回流1h,取出,放冷,再称定重量,用体积浓度为70%的甲醇溶液补足减失的重量,摇匀,滤过,精密吸取续滤液50ml,蒸干,残渣加水20ml使溶解,用水饱和的正丁醇振摇提取4次,每次25ml,取正丁醇液,蒸干,残渣加甲醇溶解并转移至5ml量瓶中,加甲醇稀释至刻度,摇匀,滤过,取续滤液,即得6份石斛夜光丸(水蜜丸)供试品溶液,各精密吸取10μl,注入液相色谱仪,测定峰面积,计算含量,结果见表6。
取石斛夜光丸(大蜜丸),剪碎,取约9g,共取6份,精密称定,置具塞锥形瓶中,加入体积浓度为70%的甲醇溶液100ml,称定重量,加热回流1h,取出,放冷,再称定重量,用体积浓度为70%的甲醇溶液补足减失的重量,摇匀,滤过,精密吸取续滤液50ml,蒸干,残渣加水20ml使溶解,用水饱和的正丁醇振摇提取4次,每次25ml,取正丁醇液,蒸干,残渣加甲醇溶解并转移至5ml量瓶中,加甲醇稀释至刻度,摇匀,滤过,取续滤液,即得6份石斛夜光丸(大蜜丸)供试品溶液,各精密吸取10μl,注入液相色谱仪,测定峰面积,计算含量,结果见表7。
结果显示,石斛夜光丸(水蜜丸)和石斛夜光丸(大蜜丸)各6份样品测得的柚皮苷、新橙皮苷、人参皂苷rg1、人参皂苷re、人参皂苷rb1含量的rsd均小于3%,符合方法学要求,表明本发明的方法重复性好。
3、精密度考察
取重复性考察中的同一份石斛夜光丸(水蜜丸)供试品溶液,重复进样6次,测定峰面积,计算rsd;另外取重复性考察中的同一份石斛夜光丸(大蜜丸)供试品溶液,重复进样6次,测定峰面积,计算rsd。结果显示,柚皮苷、新橙皮苷、人参皂苷rg1、人参皂苷re、人参皂苷rb1的峰面积rsd均小于3%,符合方法学要求,表明本发明的方法精密度好,见表8、9。
4、稳定性考察
取重复性考察中的同一份石斛夜光丸(水蜜丸)供试品溶液,分别于0h、4h、8h、16h、20h、24h进样,测定峰面积,计算rsd;另外取重复性考察中的同一份石斛夜光丸(大蜜丸)供试品溶液,分别于0h、4h、8h、16h、20h、24h进样,测定峰面积,计算rsd。结果显示,石斛夜光丸(水蜜丸)供试品溶液和石斛夜光丸(大蜜丸)供试品溶液中人参皂苷rg1、人参皂苷re、人参皂苷rb1、新橙皮苷、柚皮苷在24h内测得的峰面积rsd均小于3%,说明供试品溶液在24h内稳定,见表10、11。
6、回收率试验
(1)石斛夜光丸(水蜜丸)
1)精密称取21.37mg人参皂苷rg1,置10ml量瓶,加70%甲醇使溶解,并稀释至刻度,作为人参皂苷rg1对照品储备液(浓度为1.996mg/ml)。
2)精密称取21.79mg人参皂苷re,置10ml量瓶,加70%甲醇使溶解,并稀释至刻度,作为人参皂苷re对照品储备液(浓度为2.046mg/ml)。
3)分别精密称取27.08mg柚皮苷、20.95mg新橙皮苷、10.97mg人参皂苷rb1,置同一500ml量瓶,精密加入人参皂苷rg1对照品储备液与人参皂苷re对照品储备液各3.00ml,加70%甲醇使溶解,并稀释至刻度,作为加样用对照品混合溶液①(柚皮苷:49.665μg/ml;新橙皮苷:41.649μg/ml;人参皂苷rg1:11.976μg/ml;人参皂苷re:12.276μg/ml;人参皂苷rb1:20.426μg/ml)。
4)取石斛夜光丸(水蜜丸)适量,研碎,取约3g,精密称定,精密加入上述加样用对照品混合溶液①50ml,再精密加入70%甲醇50ml,称定重量,加热回流1h,取出,放冷,再称定重量,用体积浓度为70%的甲醇溶液补足减失的重量,摇匀,滤过,精密吸取续滤液50ml,蒸干,残渣加水20ml使溶解,用水饱和的正丁醇振摇提取4次,每次25ml,取正丁醇液,蒸干,残渣加甲醇溶解并转移至5ml量瓶中,加甲醇稀释至刻度,摇匀,滤过,取续滤液,即得加样石斛夜光丸(水蜜丸)样品溶液。
5)精密吸取加样石斛夜光丸(水蜜丸)样品溶液10μl,注入液相色谱仪,测定峰面积,计算回收率,结果加样石斛夜光丸(水蜜丸)样品溶液中柚皮苷、新橙皮苷、人参皂苷rg1、人参皂苷re、人参皂苷rb1的回收率良好,符合方法学要求,见表12。
(2)石斛夜光丸(大蜜丸)
1)精密称取21.37mg人参皂苷rg1,置10ml量瓶,加70%甲醇使溶解,并稀释至刻度,作为人参皂苷rg1对照品储备液(浓度为1.996mg/ml)。
2)精密称取21.79mg人参皂苷re,置10ml量瓶,加70%甲醇使溶解,并稀释至刻度,作为人参皂苷re对照品储备液(浓度为2.046mg/ml)。
3)精密称取33.07mg柚皮苷、20.07mg新橙皮苷、16.19mg人参皂苷rb1,置同一500ml量瓶,精密加入人参皂苷rg1对照品储备液与人参皂苷re对照品储备液各4.00ml,加70%甲醇使溶解,并稀释至刻度,作为加样用对照品溶液②(柚皮苷:56.982μg/ml;新橙皮苷:39.899μg/ml;人参皂苷rg1:15.968μg/ml;人参皂苷re:16.368μg/ml;人参皂苷rb1:30.146μg/ml)。
4)石斛夜光丸(大蜜丸)适量,研碎,取约4.5g,精密称定,精密加入加样用对照品溶液②50ml,再精密加入70%甲醇,称定重量,加热回流1h,取出,放冷,再称定重量,用体积浓度为70%的甲醇溶液补足减失的重量,摇匀,滤过,精密吸取续滤液50ml,蒸干,残渣加水20ml使溶解,用水饱和的正丁醇振摇提取4次,每次25ml,取正丁醇液,蒸干,残渣加甲醇溶解并转移至5ml量瓶中,加甲醇稀释至刻度,摇匀,滤过,取续滤液,即得加样石斛夜光丸(大蜜丸)样品溶液。
5)精密吸取加样石斛夜光丸(大蜜丸)样品溶液10μl,注入液相色谱仪,测定峰面积,计算回收率,结果加样石斛夜光丸(大蜜丸)样品溶液中柚皮苷、新橙皮苷、人参皂苷rg1、人参皂苷re、人参皂苷rb1的回收率良好,符合方法学要求,见表13。
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!