一种盐酸米那普仑中二乙胺的检测方法与流程
1.本发明涉及药物分析技术领域,尤其涉及一种盐酸米那普仑中二乙胺的检测方法。
背景技术:
2.盐酸米那普仑(milnacipran hydrochloride)是一种新型的特异性5-羟色胺和去甲肾上腺素再摄取抑制剂(snri),由法国pierre fabre medicament公司研制开发,并于1997年上市的一种治疗抑郁症的药物,2009年获得美国fda批准用于治疗纤维肌痛综合症。
3.盐酸米那普仑可同时抑制神经元对5-羟色胺和去甲肾上腺素的再摄取,从而使突触间隙的递质浓度增高,促进突触传递功能而发挥抗抑郁作用。米那普仑对脑内5-羟色胺受体及去甲肾上腺素受体具有高亲和力,可明显增加脑细胞外5-羟色胺受体及去甲肾上腺素的浓度,而对α-肾上腺素受体、毒蕈碱受体和h1组胺受体无亲和力,对单胺氧化酶活性也没有影响。由于其不参与细胞色素p450酶的反应,因而与其他药物很少发生相互作用,耐受性较好,即使长期持久性用药,也没有发现受体调节的异常现象。与三环类抗抑郁药相比,盐酸米那普仑的抗抑郁效果更加显著,明显优于安慰剂,且无三环类药物常见的嗜睡和抗胆碱作用。盐酸米那普仑不仅可以用于抑郁症的急性期治疗和维持期治疗,还可以用于治疗中风后抑郁、脑外伤后抑郁以及慢性疼痛综合征,是一种十分有前景的抗抑郁药。
4.盐酸米那普仑化学名称为:顺式-(
±
)-2-(氨基甲基)-n,n-二乙基-1-苯基环丙烷甲酰胺盐酸盐,cas:101152-94-7,分子式为:c
15h22
n2o
·
hcl,化学结构如下:
[0005][0006]
二乙胺是合成盐酸米那普仑的关键物料,容易在盐酸米那普仑原料药和制剂中残留。此外,盐酸米那普仑在储存过程中容易发生降解,产生二乙胺。二乙胺属于易挥发物质,易溶于水,具备腐蚀性及毒性,其存在会严重影响盐酸米那普仑的质量、疗效以及用药安全性,需要进行严格的质量控制。
[0007]
二乙胺的分子式为c4h
11
n,cas号:109-89-7,结构如下:
[0008][0009]
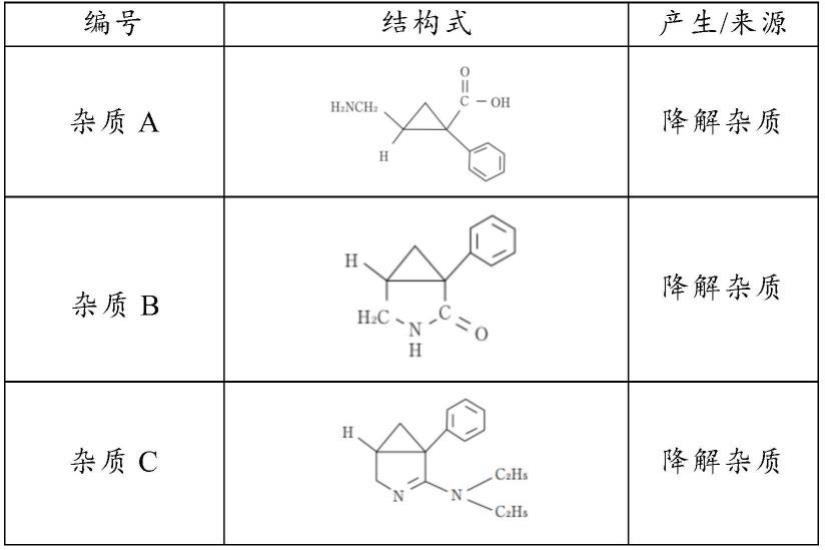
二乙胺分子极性较大,在普通硅胶色谱柱中保留差,且无紫外吸收,因此不能用普通液相色谱方法进行检测。此外,盐酸米那普仑及其制剂在放置过程中,可能会产生其他降解杂质,结构如下表1所示:
[0010]
表1盐酸米那普仑杂质
[0011][0012]
要准确检测盐酸米那普仑中残留二乙胺的含量,除了需要避免在检测过程中盐酸米那普仑发生降解,还必须使二乙胺与降解杂质a、b、c很好地分离,排除上述降解杂质对二乙胺的干扰。
[0013]
目前,尚无盐酸米那普仑中二乙胺的检测方法报道。为了有效控制盐酸米那普仑的质量,亟需开发一种盐酸米那普仑中二乙胺的有效分析检测方法。
技术实现要素:
[0014]
有鉴于此,本发明要解决的技术问题在于提供一种盐酸米那普仑中二乙胺的检测方法,可以将二乙胺与盐酸米那普仑及其他降解杂质有效分离,并定量检测二乙胺含量,分离度高。
[0015]
为达到上述目的,本发明提供了一种盐酸米那普仑中二乙胺的检测方法,包括以下步骤:
[0016]
s1)取盐酸米那普仑待测样品,进行衍生化;
[0017]
s2)采用高效液相色谱法检测,检测条件为:
[0018]
固定相:以十八烷基硅烷键合硅胶为填充剂;
[0019]
流动相:以缓冲盐溶液、乙腈、甲醇的混合溶液为流动相。
[0020]
本发明优选的,所述检测方法,包括以下步骤:
[0021]
s11)取盐酸米那普仑待测样品与衍生化试剂进行衍生化反应,制备得到供试品溶液;
[0022]
取二乙胺与衍生化试剂进行衍生化反应,制备得到对照品溶液;
[0023]
s22)采用高效液相色谱法分别对供试品溶液和对照品溶液进行检测,记录色谱图,计算待测样品中二乙胺含量。
[0024]
本发明优选的,所述缓冲盐溶液选自乙酸铵溶液。
[0025]
更优选的,所述乙酸铵溶液的浓度为1.54g/l。
[0026]
本发明优选的,所述流动相为1.54g/l乙酸铵溶液-乙腈-甲醇体系。
[0027]
所述1.54g/l乙酸铵溶液、乙腈和甲醇的体积比优选为50~54:38~42:8;更优选为52:40:8。
[0028]
本发明优选的,所述固定相为agilent infinity lab poroshell120ec-c18,填料粒径优选为2.7μm。
[0029]
本发明优选的,所述衍生化试剂为可以与二乙胺反应生成酰胺类化合物的试剂。
[0030]
更优选的,所述衍生化试剂选自苯甲酰氯。
[0031]
更优选的,所述衍生化试剂为苯甲酰氯的乙腈溶液;
[0032]
所述苯甲酰氯的乙腈溶液的浓度优选为1.8~2.5mg/ml。
[0033]
本发明优选的,所述高效液相色谱法中,检测波长为273~277nm;更优选为275nm。
[0034]
流速优选为0.7~0.9ml/min;更优选为0.8ml/min。
[0035]
柱温优选为20~30℃;更优选为25℃。
[0036]
进样量优选为10μl。
[0037]
本发明中,所述盐酸米那普仑中二乙胺的含量按峰面积外标法计算,计算公式如下:
[0038][0039]
其中,
[0040]ax
:供试品溶液中二乙胺峰峰面积;
[0041]gr
:对照品溶液中二乙胺对照品称样量,mg;
[0042]vr
:对照品溶液稀释倍数;
[0043]
p:二乙胺对照品的纯度,%;
[0044]ar
:对照品溶液中主峰平均峰面积;
[0045]gx
:供试品称样量,mg;
[0046]vx
:供试品溶液稀释倍数。
[0047]
本发明所用二乙胺对照品纯度为99.0%。
[0048]
申请人尝试采用气相色谱法检测盐酸米那普仑中二乙胺含量。结果发现,盐酸米那普仑在气相色谱条件下不稳定,容易降解产生二乙胺,对盐酸米那普仑中的二乙胺实际含量检测造成干扰。因此,气相色谱检测法无法准确检测盐酸米那普仑中的二乙胺含量。
[0049]
与现有技术相比,本发明提供了一种盐酸米那普仑中二乙胺的检测方法,包括以下步骤:s1)取盐酸米那普仑待测样品,进行衍生化;s2)采用高效液相色谱法检测,检测条件为:固定相:以十八烷基硅烷键合硅胶为填充剂;流动相:以缓冲盐溶液、乙腈、甲醇的混合溶液为流动相。
[0050]
本发明提供的检测方法可以对盐酸米那普仑中二乙胺进行有效分离并定量测定,同时避免了盐酸米那普仑在检测过程中发生降解产生二乙胺而影响二乙胺的准确检测,为精确检测二乙胺的含量提供了一种有效的方法,从而有效控制盐酸米那普仑原料药和制剂的质量。本发明提供的检测方法基线无干扰,分离度高,检测限低,专属性强,灵敏度高,准确度、精密度及耐用性好。
[0051]
同时,本发明提供的分析检测方法操作简单,条件温和,对高效液相色谱仪器、分
析试剂和色谱柱没有苛刻要求,能够适用于一般的色谱柱和高效液相分析仪器。
附图说明
[0052]
图1为比较例1中二乙胺对照溶液色谱图;
[0053]
图2为比较例1中杂质加样溶液色谱图;
[0054]
图3为比较例3中衍生化溶液a的色谱图;
[0055]
图4为比较例3中衍生化溶液b的色谱图;
[0056]
图5为实施例3中空白溶液色谱图;
[0057]
图6为实施例3中二乙胺对照溶液色谱图;
[0058]
图7为实施例3中供试品溶液色谱图;
[0059]
图8为实施例4专属性试验中分离度测试溶液色谱图。
具体实施方式
[0060]
为了进一步说明本发明,下面结合实施例对本发明提供的盐酸米那普仑中二乙胺的检测方法进行详细描述。
[0061]
本发明具体实施方式中使用的原料、试剂和仪器均为已知产品,可通过市售购买获得;盐酸米那普仑按照已有方法制备。
[0062]
比较例1
[0063]
(1)色谱条件(顶空进样turbo matrix 40)
[0064]
仪器:气相色谱仪
[0065]
检测器:fid检测器
[0066]
色谱柱:cp-volamine(30m
×
0.32mm),agilent
[0067]
升温程序:起始温度40℃,维持10分钟,再以15℃每分钟升至180℃,维持10分钟
[0068]
进样口:200℃
[0069]
检测口:250℃
[0070]
载气:n2,1ml/min
[0071]
分流比:10:1
[0072]
顶空条件:平衡温度:90℃,平衡时间:30分钟
[0073]
进样量:0.04min,加压时间:2min
[0074]
溶剂:n,n-二甲基甲酰胺
[0075]
(2)溶液配制
[0076]
空白溶液:取n,n-二甲基甲酰胺5ml于20ml顶空瓶内,密封,即得。
[0077]
二乙胺对照溶液:取二乙胺20mg,精密称定,置20ml容量瓶内,用n,n-二甲基甲酰胺稀释至刻度,混匀;再量取该溶液1ml于50ml容量瓶内,用n,n-二甲基甲酰胺稀释至刻度,混匀,取5ml于20ml顶空瓶内,密封,即得。
[0078]
供试品溶液:取盐酸米那普仑500mg,精密称定,置20ml顶空瓶内,加5ml n,n-二甲基甲酰胺溶解,混匀,密封,即得。
[0079]
杂质加样溶液:取盐酸米那普仑500mg,精密称定,置20ml顶空瓶内,加5ml二乙胺对照溶液溶解,混匀,密封,即得。
[0080]
(3)测定
[0081]
按上述色谱条件试验,取空白溶液、二乙胺对照溶液、供试品溶液、杂质加样溶液分别进样,记录色谱图。
[0082]
(4)检测结果
[0083]
典型色谱图见图1~图2。从图谱结果看,二乙胺对照溶液色谱图中,二乙胺峰形与响应较好,但在杂质加样溶液中未检出等量的二乙胺。分析原因可能与盐酸米那普仑偏酸性有关,使二乙胺无法完全游离出来。因此,考虑加入氢氧化钠碱化溶液,让二乙胺游离,再通过fid检测器进行检测。
[0084]
比较例2
[0085]
1)色谱条件(顶空进样turbo matrix 40)
[0086]
仪器:气相色谱仪
[0087]
检测器:fid检测器
[0088]
色谱柱:cp-volamine(30m
×
0.32mm),agilent
[0089]
升温程序:起始温度60℃,维持7分钟,再以10℃每分钟升至220℃,维持7分钟
[0090]
进样口:140℃
[0091]
检测口:200℃
[0092]
载气:n2,1ml/min
[0093]
分流比:2:1
[0094]
顶空条件:炉温:60℃,定量环温度:60℃
[0095]
传输线温度:75℃,平衡时间:25分钟
[0096]
进样量:0.1min
[0097]
溶剂:10mg/ml naoh溶液
[0098]
(2)溶液配制
[0099]
空白溶液(即10mg/ml naoh溶液):称取氢氧化钠2.0g,加200ml新沸冷水溶解并稀释,混匀取5ml于20ml顶空瓶内,密封,即得。
[0100]
二乙胺对照溶液:二乙胺27.5mg,精密称定,置10ml容量瓶内,用10mg/ml naoh溶液稀释至刻度,混匀;再量取该溶液1ml于100ml容量瓶内,用10mg/ml naoh溶液稀释至刻度,混匀,取5ml于20ml顶空瓶内,密封,即得。
[0101]
供试品溶液:取盐酸米那普仑500mg,精密称定,置20ml顶空瓶内,加5ml 10mg/ml naoh溶液溶解,混匀,密封,即得。
[0102]
杂质加样溶液:取盐酸米那普仑500mg,精密称定,置20ml顶空瓶内,加5ml二乙胺对照溶液溶解,混匀,密封,即得。
[0103]
(3)测定
[0104]
按上述色谱条件试验,取空白溶液、二乙胺对照溶液、供试品溶液、杂质加样溶液分别进样,记录色谱图。
[0105]
(4)检测结果
[0106]
该色谱条件下,二乙胺检出情况如表2所示:
[0107]
表2二乙胺检出情况
[0108][0109]
由表2可知,在优化了溶液配制方法后,可以检出盐酸米那普仑中的二乙胺。但是,常温下放置15h后,供试品溶液和杂质加样溶液中二乙胺的峰面积明显增大,说明在碱性条件下,盐酸米那普仑不稳定,发生降解,产生二乙胺。因为盐酸米那普仑降解产生二乙胺的影响,该气相色谱方法不能准确检测盐酸米那普仑中残留二乙胺的含量。
[0110]
比较例3
[0111]
(1)色谱条件
[0112]
仪器:高效液相色谱仪agilent 1260
[0113]
色谱柱:agilent poroshell 120 ec-c18,4.6
×
100mm,2.7μm
[0114]
流动相:a相:1.54g/l乙酸铵水溶液,用氨水调ph为7.0
[0115]
b相:乙腈
[0116]
c相:甲醇
[0117]
流速:0.8ml/min
[0118]
波长:263nm
[0119]
柱温:25℃
[0120]
进样量:10μl
[0121]
进样器温度5℃
[0122]
洗脱条件:
[0123]
表3洗脱条件
[0124][0125]
(2)溶液配制
[0126]
四硼酸钠溶液:50mg/ml四硼酸钠溶液、乙腈、超纯水按5:15:7(体积比)比例混合,即得。
[0127]
衍生化试剂1:称取50mg苯甲酰氯,溶解于25ml乙腈中,摇匀,即得。
[0128]
衍生化试剂2:称取50mg芴甲氧羰酰氯,溶解于25ml乙腈中,摇匀,即得。
[0129]
空白溶液
①
:取纯化水250μl于棕色瓶中,加入1350μl四硼酸钠溶液,混匀,放置8min,再量取该溶液750μl与750μl衍生化试剂1于棕色样品瓶中,混合均匀,25℃放置1h即得。
[0130]
空白溶液
②
:取纯化水250μl于棕色瓶中,加入1350μl四硼酸钠溶液,混匀,放置8min,再量取该溶液750μl与750μl衍生化试剂2于棕色样品瓶中,混合均匀,25℃放置1h即得。
[0131]
二乙胺储备液:取二乙胺10mg,精密称定,置10ml容量瓶内,用乙腈稀释至刻度,混匀,即得。
[0132]
衍生化溶液a:取二乙胺储备液250μl,置棕色样品中,加入1350μl四硼酸钠溶液,混合均匀,放置8分钟,量取该溶液750μl,置棕色样品瓶中,加入750μl衍生化试剂1,混合均匀,25℃放置1小时后,即得。
[0133]
衍生化溶液b:取二乙胺储备液250μl,置棕色样品中,加入1350μl四硼酸钠溶液,混合均匀,放置8分钟,量取该溶液750μl,置棕色样品瓶中,加入750μl衍生化试剂2,混合均匀,25℃放置1小时后,即得。
[0134]
(3)测定
[0135]
按上述色谱条件试验,取空白溶液
①
、空白溶液
②
、衍生化溶液a、衍生化溶液b分别进样,记录色谱图。
[0136]
(4)测定结果
[0137]
典型色谱图见图3~图4。由图可知,以苯甲酰氯作为衍生化试剂的二乙胺衍生化峰,在液相色谱条件下响应优于芴甲氧羰酰氯作衍生化试剂时的响应。
[0138]
实施例1
[0139]
方法一
[0140]
(1)色谱条件
[0141]
仪器:高效液相色谱仪agilent 1260
[0142]
色谱柱:agilent poroshell 120 ec-c18,4.6
×
100mm,2.7μm
[0143]
流动相:a相:1.54g/l乙酸铵水溶液,用氨水调ph为7.0
[0144]
b相:乙腈
[0145]
c相:甲醇
[0146]
流速:0.8ml/min
[0147]
波长:263nm
[0148]
柱温:25℃
[0149]
进样量:10μl
[0150]
进样器温度5℃
[0151]
洗脱条件:a相:b相:c相=52:40:8
[0152]
(2)溶液配制
[0153]
四硼酸钠溶液:同比较例3。
[0154]
衍生化试剂1:同比较例3。
[0155]
空白溶液
①
:同比较例3。
[0156]
二乙胺储备液:同比较例3。
[0157]
衍生化溶液a:同比较例3。
[0158]
(3)测定
[0159]
按上述色谱条件试验,取空白溶液
①
、衍生化溶液a分别进样,记录色谱图。
[0160]
方法二
[0161]
洗脱条件:a相:b相:c相=40:52:8,其他条件同方法一。
[0162]
方法三
[0163]
洗脱条件:a相:b相:c相=32:60:8,其他条件同方法一。
[0164]
(4)测定结果
[0165]
方法一、二、三中,区别点在于洗脱条件不同。当采用方法一的洗脱条件时,基线波动最小,且二乙胺衍生化峰与各空白峰分离度较好。故选取方法一继续进行检测方法开发。
[0166]
实施例2
[0167]
方法四
[0168]
(1)色谱条件
[0169]
仪器:高效液相色谱仪agilent 1260
[0170]
色谱柱:agilent poroshell 120 ec-c18,4.6
×
100mm,2.7μm
[0171]
流动相:a相:1.54g/l乙酸铵水溶液,用氨水调ph为7.0
[0172]
b相:乙腈
[0173]
c相:甲醇
[0174]
流速:0.8ml/min
[0175]
波长:265nm
[0176]
柱温:25℃
[0177]
进样量:10μl
[0178]
进样器温度:5℃
[0179]
洗脱条件:a相:b相:c相=52:40:8
[0180]
(2)溶液配制
[0181]
10mg/ml磷酸氢二钠溶液:称取5.0g无水磷酸氢二钠,溶解在500ml水中,摇匀,即得。
[0182]
衍生化试剂:称取50mg苯甲酰氯,溶解于25ml乙腈中,摇匀,即得。
[0183]
空白溶液:取纯水250μl,置棕色样瓶中,加入1350μl 10mg/ml磷酸氢二钠溶液,混匀,放置8min;取该溶液750μl于棕色样瓶中,加入750μl衍生化试剂,混匀,常温下放置1小时,即得;
[0184]
二乙胺储备液:取二乙胺56mg,精密称定,置10ml量瓶内,用乙腈稀释至刻度,混匀;取该溶液1ml,置10ml量瓶内,用水稀释至刻度,混匀,即得;
[0185]
杂质加样溶液:取盐酸米那普仑10mg,精密称定,置10ml量瓶内,加二乙胺储备液1ml,再用水溶解并稀释至刻度,混匀,即得;
[0186]
衍生化杂质加样溶液:取杂质加样溶液250μl,置棕色样瓶中,加入1350μl10mg/ml磷酸氢二钠溶液,混匀,放置8min;取该溶液750μl于棕色样瓶中,加入750μl衍生化试剂,混匀,常温下放置1小时,即得;
[0187]
(3)测定
[0188]
按上述色谱条件试验,取空白溶液、衍生化杂质加样溶液分别进样,记录色谱图。
[0189]
方法五
[0190]
波长:267nm,其他条件同方法四。
[0191]
方法六
[0192]
波长:275nm,其他条件同方法四。
[0193]
(4)测定结果
[0194]
分别采用方法四、五、六进行检测,结果发现,当检测波长为275nm时,空白在二乙胺衍生化峰处无干扰。故优选275nm波长。
[0195]
实施例3盐酸米那普仑中二乙胺检测
[0196]
(1)色谱条件
[0197]
仪器:高效液相色谱仪agilent 1260;
[0198]
色谱柱:agilent infinity lab poroshell 120 ec-c18,4.6
×
100mm,2.7μm;
[0199]
流动相:1.54g/l乙酸铵溶液-乙腈-甲醇=52:40:8;
[0200]
流速:0.8ml/min;
[0201]
检测波长:275nm;
[0202]
柱温:25℃;
[0203]
进样体积:10μl;
[0204]
运行时间:25min。
[0205]
(2)溶液配制
[0206]
10mg/ml磷酸氢二钠溶液:称取5.0g无水磷酸氢二钠,溶解在500ml水中,摇匀,即得。
[0207]
衍生化试剂:称取50mg苯甲酰氯,溶解于25ml乙腈中,摇匀,即得。
[0208]
空白溶液:取纯水3ml,置20ml容量瓶中,用10mg/ml磷酸氢二钠溶液稀释至刻度,摇匀;量取该溶液500μl,置棕色样品瓶中,加入500μl衍生化试剂,混合均匀,常温放置10分钟,即得。
[0209]
二乙胺对照溶液:精密称取二乙胺110mg,置10ml容量瓶中,加水稀释至刻度,摇匀;精密量取该溶液1.0ml,置100ml容量瓶中,加水稀释至刻度,摇匀;精密量取该溶液1.0ml,置10ml容量瓶中,加水稀释至刻度,摇匀,精密量取3ml,置20ml容量瓶中,用10mg/ml磷酸氢二钠溶液稀释至刻度,摇匀;量取该溶液500μl,置棕色样品瓶中,加入500μl衍生化试剂,混合均匀,常温放置10分钟,即得。
[0210]
供试品溶液:精密称取盐酸米那普仑样品200mg,置10ml容量瓶中,加水溶解并稀释至刻度,摇匀,再精密量取3ml,置20ml容量瓶中,用10mg/ml磷酸氢二钠溶液稀释至刻度,摇匀;量取该溶液500μl,置棕色样品瓶中,加入500μl衍生化试剂,混合均匀,常温放置10分钟,即得。
[0211]
(3)测定
[0212]
按上述色谱条件试验,取空白溶液、二乙胺对照溶液、供试品溶液分别进样,记录色谱图。
[0213]
(4)测定结果
[0214]
典型色谱图见图5~图7。图6中保留时间为2.929min的色谱峰为二乙胺衍生化后色谱峰。图7中保留时间为8.185min的色谱峰为盐酸米那普仑样品衍生化后色谱峰。由图可知,空白溶剂不干扰测定,所测盐酸米那普仑样品中未检出二乙胺残留。
[0215]
实施例4方法验证
[0216]
对实施例3的检测方法进行验证,以证明本发明的技术效果。
[0217]
1、专属性
[0218]
10mg/ml磷酸氢二钠溶液:称取5.0g无水磷酸氢二钠,溶解在500ml水中,摇匀,即得。
[0219]
衍生化试剂:称取50mg苯甲酰氯,溶解于25ml乙腈中,摇匀,即得。
[0220]
空白溶液:取纯水3ml,置20ml容量瓶中,用10mg/ml磷酸氢二钠溶液稀释至刻度,摇匀;量取该溶液500μl,置棕色样品瓶中,加入500μl衍生化试剂,混合均匀,常温放置10分钟,即得。
[0221]
二乙胺贮备液:精密称取二乙胺110mg,置10ml容量瓶中,加水稀释至刻度,摇匀;精密量取该溶液1.0ml,置100ml容量瓶中,加水稀释至刻度,摇匀。
[0222]
二乙胺对照溶液:精密量取二乙胺贮备液1.0ml,置10ml容量瓶中,加水稀释至刻度,摇匀,精密量取3ml,置20ml容量瓶中,用10mg/ml磷酸氢二钠溶液稀释至刻度,摇匀;量取该溶液500μl,置棕色样品瓶中,加入500μl衍生化试剂,混合均匀,常温放置10分钟,即得。
[0223]
杂质a贮备液:称取20mg杂质a,置10ml量瓶内,加水溶解并稀释至刻度,摇匀,即得。
[0224]
杂质b贮备液:称取20mg杂质b,置10ml量瓶内,加水溶解并稀释至刻度,摇匀,即得。
[0225]
杂质c贮备液:称取20mg杂质c,置10ml量瓶内,加水溶解并稀释至刻度,摇匀,即得。
[0226]
杂质a定位溶液:精密移取杂质a贮备液1.0ml置10ml量瓶中,加水稀释至刻度,即得。
[0227]
杂质b定位溶液:精密移取杂质b贮备液1.0ml置10ml量瓶中,加水稀释至刻度,即得。
[0228]
杂质c定位溶液:精密移取杂质c贮备液1.0ml置10ml量瓶中,加水稀释至刻度,即得。
[0229]
衍生化杂质a定位溶液:精密移取杂质a贮备液3.0ml置20ml容量瓶中,用10mg/ml磷酸氢二钠溶液稀释至刻度,摇匀;量取该溶液500μl,置棕色样品瓶中,加入500μl衍生化试剂,混合均匀,常温放置10分钟,即得;
[0230]
衍生化杂质b定位溶液:精密移取杂质b贮备液3.0ml置20ml容量瓶中,用10mg/ml磷酸氢二钠溶液稀释至刻度,摇匀;量取该溶液500μl,置棕色样品瓶中,加入500μl衍生化试剂,混合均匀,常温放置10分钟,即得;
[0231]
衍生化杂质c定位溶液:精密移取杂质c贮备液3.0ml置20ml容量瓶中,用10mg/ml磷酸氢二钠溶液稀释至刻度,摇匀;量取该溶液500μl,置棕色样品瓶中,加入500μl衍生化试剂,混合均匀,常温放置10分钟,即得;
[0232]
100%限度杂质加样溶液:取盐酸米那普仑200mg,置10ml容量瓶内,加适量水溶解,再分别加入二乙胺贮备液、杂质a贮备液、杂质b贮备液、杂质c贮备液各1.0ml,用水稀释至刻度,摇匀;量取3ml,置20ml容量瓶中,用10mg/ml磷酸氢二钠溶液稀释至刻度,摇匀;量取该溶液500μl,置棕色样品瓶中,加入500μl衍生化试剂,混合均匀,常温放置10分钟,即
得。
[0233]
通过空白溶液的干扰、100%限度杂质加样溶液中相邻杂质分离度来评价方法的专属性。
[0234]
其中,杂质a、b、c如表1所示。
[0235]
测试结果如表4、表5及图8所示:
[0236]
表4各杂质单独定位结果
[0237][0238]
表5分离度测试结果
[0239][0240]
结果表明,空白溶液基线平稳且无干扰;100%限度杂质加样溶液中二乙胺与相邻杂质峰之间分离度最小为8.58,符合要求。
[0241]
2、系统适用性
[0242]
连续6针二乙胺对照溶液(配制方法同专属性试验中二乙胺对照溶液配制方法)进样,测定二乙胺峰面积的rsd,二乙胺峰面积的rsd应小于3.0%。
[0243]
结果如表6所示。
[0244]
表6二乙胺对照溶液检测结果
[0245][0246]
结果表明,连续6针二乙胺对照溶液,峰面积rsd为1.2%,符合要求。
[0247]
3、定量限
[0248]
接受标准:5针定量限溶液的信噪比(s/n)值应大于9;5针定量限溶液中峰面积的rsd不得过15%;定量限浓度应不大于50%限度浓度。
[0249]
验证结果:定量限溶液的信噪比(s/n)值均大于9,峰面积rsd为9.7%,定量限浓度约相当于限度浓度的9%(限度为550ppm),符合要求。
[0250]
4、检测限
[0251]
接受标准:检测限溶液的信噪比(s/n)值应大于2。
[0252]
验证结果:检测限溶液的信噪比(s/n)值均大于4.3,符合要求。
[0253]
5、线性
[0254]
在定量限浓度至200%限度浓度范围内取5个浓度点进行研究,以测得的响应信号(峰面积)对被测物的浓度作图,进行线性回归,要求该曲线的线性回归相关系数(r)的数值应不小于0.999,y轴截距应不得大于限度浓度响应值的10%。
[0255]
验证结果:二乙胺线性方程为y=1.4719x+0.0531,相关系数r为0.9998;y轴截距为0.0531,相当于限度浓度响应值的4.2%,符合要求。
[0256]
6、准确度
[0257]
准确度是通过在供试品溶液中加入定量限浓度、100%限度浓度和120%限度浓度三个不同浓度各杂质对照品测得的各杂质的回收率评价。杂质的准确度是加入已知量的杂质,再测定加样样品中已知杂质的测定结果和理论值之间的比值(回收率),以百分比表达,要求回收率应在85%~110%之间,回收率rsd应不得过10%。
[0258]
结果:定量限准确度回收率为104.3%,100%准确度回收率为92.7%,120%准确度回收率为91.2%,回收率rsd为6.8%。符合要求。
[0259]
7、精密度
[0260]
1)重复性
[0261]
重复性是通过配制6份加样溶液进行测试,每个溶液进样1针来验证的。要求6次测定结果的rsd应不大于10.0%。
[0262]
结果:重复性结果rsd(n=6)为3.0%,符合要求。
[0263]
2)中间精密度
[0264]
为考察随机变动对精密度的影响,分别由2名分析人员在不同日期对同一批次样品进行加样测试,每位分析人员测试6份。以12份测定结果(二乙胺含量)的相对标准偏差rsd来评价中间精密度,要求12份测定结果的rsd应不大于10.0%。
[0265]
结果:由两位分析员在不同时间对同一批次样品进行检测,共计12份样品加标溶液中二乙胺含量的rsd(n=12)为2.6%,符合要求。
[0266]
8、范围
[0267]
结果:二乙胺浓度在0.0816μg/ml~1.6322μg/ml范围内,精密度、准确度、线性关系良好。
[0268]
9、耐用性
[0269]
1)溶液稳定性
[0270]
考察二乙胺对照溶液、杂质加样溶液随时间变化的规律,分别在一定时间内考察杂质峰面积的变化情况。对照溶液中二乙胺峰面积与0小时峰面积比较,相对偏差不大于10.0%,则对照溶液不用临用新制,反之则对照溶液需临用新制。杂质加样溶液中二乙胺峰面积与0小时峰面积比较,若相对偏差不大于10.0%,则样品溶液不用临用新制,反之则供试品溶液需临用新制。
[0271]
结果:二乙胺对照溶液放置38h后,相对偏差为2.25%,杂质加样溶液放置38h后,相对偏差为5.54%,相对偏差均小于10%,表明二乙胺对照溶液与杂质加样溶液在38h内均稳定。
[0272]
2)色谱条件与衍生化条件改变
[0273]
通过改变流速
±
0.1ml/min、柱温
±
5℃、波长
±
2nm、流动相比例调节(1.54g/l乙
酸铵溶液-乙腈-甲醇=50:42:8~54:38:8)、磷酸氢二钠浓度
±
1mg/ml、苯甲酰氯浓度
±
0.5mg/ml来评估色谱条件的承受程度与衍生化程度。要求杂质加样溶液与二乙胺对照溶液中已知杂质峰与相邻杂质分离度应不小于1.5,在各变动色谱条件下,加标样品溶液中二乙胺含量的rsd不得过10%。
[0274]
结果:
[0275]
色谱条件发生微小变动(流速
±
0.1ml/min、柱温
±
5℃、波长
±
2nm、流动相比例调节:1.54g/l乙酸铵溶液-乙腈-甲醇=50:42:8~54:38:8)时,杂质加样溶液中二乙胺含量的rsd最大2.7%,符合要求。
[0276]
衍生化条件中,磷酸氢二钠浓度发生微小变动时(
±
1mg/ml),杂质加样溶液中二乙胺含量的rsd为0.7%,符合要求。
[0277]
苯甲酰氯浓度发生变动(
±
0.5mg/ml)的考察中,当苯甲酰氯浓度在1.5mg/ml时,二乙胺检出量明显低于原条件,1.5mg/ml~2.5mg/ml范围内,二乙胺检出量rsd为25.6%,不符合要求;当苯甲酰氯浓度为1.8mg/ml~2.5mg/ml范围时,二乙胺检出量与原条件检出结果相当,rsd为6.7,小于10%,符合要求。
[0278]
通过对盐酸米那普仑中二乙胺的测定方法系统适用性、专属性、定量限、检测限、线性及范围、准确度、精密度和耐用性项目的验证,结果表明各项均符合要求,本发明提供的上述检测方法可用于盐酸米那普仑中二乙胺的测定,从而有效控制盐酸米那普仑及其制剂的质量,有利于保证用药的安全性和有效性。
[0279]
以上实施例的说明只是用于帮助理解本发明的方法及其核心思想。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1