一种高通量筛选Aβ纤维形成抑制剂的方法
一种高通量筛选a
β
纤维形成抑制剂的方法
技术领域
1.本发明涉及了高通量筛选技术领域,具体涉及了一种高通量筛选aβ纤维形成抑制剂的方法。
背景技术:
2.阿尔茨海默氏病的病理与不断累积的错误折叠的蛋白质聚集体(例如β
‑
淀粉样蛋白(aβ)和tau)引起的神经炎症和神经元死亡的发生密切相关。aβ是由淀粉样前体蛋白(app)通过顺序切割β
‑
和γ
‑
分泌酶产生的。所产生的包括aβ(1
‑
40)和aβ(1
‑
42)的片段被认为是毒性最高的形式,形成aβ原纤维并在细胞外积聚,最终在阿尔茨海默氏病患者的大脑中产生老年斑。在阿尔茨海默氏病的早期阶段,aβ仍可通过神经元中的自噬溶酶体途径(alp)和泛素
‑
蛋白酶体途径(upp)清除,或被小胶质细胞吞噬。但是,alp和upp均会随着年龄的增长而失调,使得aβ的清除无法有效执行。因此,靶向抑制aβ原纤维的形成已成为阿尔茨海默氏病的有希望的治疗策略。
3.研究发现许多中药或其成分在阿尔茨海默氏病的体外和体内模型中显示出有效的神经保护作用。但是从具有复杂化学成分的中草药或配方中发现生物活性成分的过程既费时又费力。迄今为止,已报道了许多针对中药的筛选方法,包括细胞膜层析(cmc),网络药理学和分子对接,与目标药物的亲和超滤以及高效液相色谱
‑
质谱联用(auf
‑
hplc
‑
ms/ms),结合lc
‑
ms的气泡产生磁性脂质体等。通常,大多数方法都是基于高级分析仪器(例如hplc,ms和核磁共振(nmr))的应用而开发的。
4.因此,开发一种有效、省时、可靠且简单的方法,从中药中筛选出aβ纤维形成抑制剂,已变得非常重要。
技术实现要素:
5.本发明的目的在于:针对现有技术从具有复杂化学成分的中草药或配方中发现具有抑制aβ纤维形成的生物活性成分的过程存在既费时又费力的问题,提供一种高通量筛选aβ纤维形成抑制剂的方法。该方法操作简单,省时,且有效,可靠,便于推广应用。
6.为了实现上述目的,本发明采用的技术方案为:
7.一种高通量筛选aβ纤维形成抑制剂的方法,包括以下步骤:
8.步骤1、生物膜干涉分析
9.制备待测中草药提取物,然后用磷酸盐缓冲液将待测中草药提取物进行稀释得到工作液;
10.制备生物素化的aβ(1
‑
42)肽;
11.将工作液和生物素化的aβ(1
‑
42)肽固定于生物传感器上,进行结合
‑
解离,得到结合和解离图、动力学常数,得到待测中草药提取物与生物素化的aβ(1
‑
42)肽的结合情况;
12.步骤2、液质联用分析
13.采用步骤1相同的方法,将待测中草药提取物与生物素化的aβ(1
‑
42)肽在生物传
感器中连续进行n次结合
‑
解离操作,收集解离液,其中n为大于或等于5的自然数;
14.然后将收集的解离液在氮气流下进行干燥处理;
15.之后,将干燥后的解离液用有机溶剂进行复溶,复溶液放入超高效液相色谱串联质谱检测仪器中,得到代表性总离子色谱图,分析谱图,筛选出具有与aβ纤维直接结合作用的化合物,即为aβ纤维形成抑制剂。
16.本发明筛选aβ纤维形成抑制剂的高通量方法主要通过生物膜干涉分析和液质联用分析结合使用的结果,本发明需要检测的是待测中草药提取物中多种为未知化合物与生物素化的aβ(1
‑
42)肽的结合情况,然后根据结合情况,设计多次重复结合
‑
解离操作,收集解离液,并对收集的解离液进行液质联用分析,根据代表性总离子色谱图分析即可筛选出与aβ纤维存在直接结合的单体。并使用后续分子生物学实验,筛选出其中有抑制作用的单体。本发明提供的高通量筛选方法,操作简单,占用时间少,准确度高,精准性高,重复性好,便于广泛的推广应用,对从中药中轻松的筛选出aβ纤维形成抑制剂具有十分重要的意义。
17.进一步的,所述步骤1中,制备待测中草药提取物过程中,提取溶剂为水、乙醇或甲醇。
18.进一步的,所述步骤1中,待测中草药提取物具体由以下方法制备而成的:将待测中草药进行称量、粉碎,然后用8
‑
12倍体积的水、乙醇或甲醇进行蒸馏提取1
‑
2h,重复提取3
‑
5次,合并提取液,过滤,浓缩,然后将浓缩后的提取物用dmso(二甲基亚砜)溶解为200~500mg/ml的溶液,得到待测中草药提取物。
19.进一步的,所述步骤1中,所述工作液浓度为25~4000μg/ml。
20.进一步的,所述步骤1中,所述生物素化的aβ(1
‑
42)肽主要由以下方法制备而成的:
21.将摩尔比为2
‑
6:1的生物素与aβ(1
‑
42)肽溶液进行混合,然后再室温下孵育30min~60min,得到生物素化的aβ(1
‑
42)肽。
22.进一步的,所述aβ(1
‑
42)肽溶液主要由以下方法制备而成的:
23.将aβ(1
‑
42)肽置于六氟异丙醇中,然后将得到的混合溶剂分装到试管中进行氮气流下干燥,得到干燥后的薄膜,然后在试管中加入dmso(二甲基亚砜)和磷酸盐缓冲剂,得到aβ(1
‑
42)肽溶液。进一步的,所述aβ(1
‑
42)肽溶液的浓度为100~200mg/ml。
24.进一步的,所述步骤1中,当结合和解离图中响应值上升说明待测中草药提取物与生物素化的aβ(1
‑
42)肽有结合,则再进行步骤2的分析。优选地,所述步骤1中,当得到的动力学常数在1
×
105内,说明待测中草药提取物与生物素化的aβ(1
‑
42)肽有结合且结合能力强,则再进行步骤2的分析。
25.进一步的,所述步骤1中,采用不同浓度的工作液进行多次结合
‑
解离,得到多个结合和解离图、动力学常数。通过不同浓度工作液进行实验,可以充分准确的判断待测中草药提取物与生物素化的aβ(1
‑
42)肽的结合情况,进一步的增加实验的稳定性和准确度。
26.进一步的,所述步骤2中,n为5
‑
10的自然数。经过发明人大量的实验研究探索,当结合
‑
解离重复操作的次数较少,低于5次时,无法达到质谱仪检测限,误差大,不精确。但是结合
‑
解离次数不能过多,当次数超过10次后,解离液中非特异性结合单体对质谱检测的干扰增加,导致检测结构误差大,稳定性差。
27.进一步的,所述超高效液相色谱串联质谱检测仪器为超高效液相色谱串联四极杆
飞行时间质谱仪器(超高效液相色谱
‑
dad
‑
q/tof
‑
ms/ms)。
28.进一步的,所述步骤2中,有机溶剂为乙醇或甲醇。
29.进一步的,所述步骤2中,超高效液相色谱分析条件为:
30.c18柱,柱温为30℃~45℃;流动性为流动相a(0.1%甲酸水溶液)和b(0.1%甲酸乙腈溶液)组成;
31.流速为0.2~0.5ml/min。
32.优选地,所述步骤2中,超高效液相色谱分析条件为:
33.zorbax eclipse plus c18柱100mm
×
2.1mm
×
1.8um;流速:0.3ml/min;柱温设定在40℃,流动相由a(0.1%甲酸水溶液)和b(0.1%甲酸乙腈溶液)组成。
34.进一步的,所述步骤2中,质谱测试的条件为:
35.duo spray源为负电喷雾离子模式,电喷雾电离按照以下参数进行:喷雾电压,
‑
4,500v;离子源温度,550℃;气帘气,35psi;雾化气(gs 1),55psi;加热器气(gs 2),55psi;解聚电位(dp),
‑
100v;ms扫描的质量范围设置为m/z 100
‑
1,600da。
36.进一步的,所述步骤2中,分析高效液相仪检测结果峰面积比值,得到所有化合物的rba值,对不同化合物rba值进行大小比较,rba值较大出现断层式增加的化合物为具有与aβ纤维直接结合作用的化合物。
37.进一步的,rba值较大出现断层式高度的化合物的判定方法为:对不同化合物rba值进行由高到低的顺序排列成第1个化合物......第n个化合物,第n+1个化合物,第n+2个化合物,第n+3个化合物.......第n+n个化合物,当第n个化合物的rba值比第n+1个化合物的rba值高出100%以上时,则第1个化合物~n个化合物为筛选出aβ纤维形成抑制剂化合物,其中n为大于或等于1的自然数,n为0或≥1的自然数。
38.优选地,当第n个化合物的rba值比第n+1个化合物的rba值高出200%以上时,则第1个化合物~n个化合物为筛选出aβ纤维形成抑制剂化合物。
39.更优选地,当第n个化合物的rba值比第n+1个化合物的rba值高出250%以上时,则第1个化合物~n个化合物为筛选出aβ纤维形成抑制剂化合物。例如300%以上,350%以上......。
40.解释文中出现缩写中文意思:
41.sb
‑
黄芩;kxs
‑
开心散;dta
‑
去氢土莫酸;ta
‑
土莫酸;ppac
‑
猪苓酸c;poria
‑
茯苓;cur
‑
姜黄素。
42.综上所述,由于采用了上述技术方案,本发明的有益效果是:
43.1.本发明提供的筛选aβ纤维形成抑制剂的高通量方法,通过生物膜干涉分析和液质联用分析组合使用,最终根据代表性总离子色谱图分析即可筛选出aβ纤维形成抑制剂化合物。该筛选方法,操作简单,占用时间少,准确度高,精准性高,重复性好,便于广泛的推广应用,对从中药中轻松的筛选出aβ纤维形成抑制剂具有十分重要的意义,加速了未来抗ad药物的发现和发展进程。
附图说明
44.图1是实施例1中不同浓度黄芩提取物动态结合和解离传感图。
45.图2是实施例1中黄芩提取物代表性总离子色谱图。
46.图3是实施例1中黄芩提取物中检测到的成分的rba的柱状图。
47.图4是实施例2中不同浓度开心散提取物动态结合和解离传感图。
48.图5是实施例2中开心散提取物代表性总离子色谱图。
49.图6是实施例2中s1、s2和s3中dta的萃取离子色谱图(eics)。
50.图7是实施例2中s1、s2和s3中的ta的eics。
51.图8是实施例2中s1、s2和s3中ppac的eics。
52.图9是实施例3中ppac、dta、ta和茯苓提取物的tic图。
53.图10是实施例3中ppac、dta和ta的质谱图。
54.图11是实施例3中ppac、dta和ta的分子结构图。
55.图12为实施例3中0h时间点各个检测物的荧光强度图。
56.图13为实施例3中24h时间点各个检测物的荧光强度图。
57.图14为实施例3中48h时间点各个检测物的荧光强度图。
58.图15为实施例3中mtt法检测各个检测物的细胞毒性反应图。
59.图16为实施例3中pc
‑
12细胞的细胞存活率百分比柱状图。
60.图17为实施例3中pc
‑
12细胞的细胞死亡百分比情况图。
61.图18为实施例3中pc
‑
12细胞的pi/hoechst信号的比率。
62.图19为实施例3中测试的aβ诱导线虫株cl4176在各检测物作用下的瘫痪速率。
63.图20为实施例3中测试的aβ诱导对照株线虫cl2122在各检测物作用下线虫感知食物速率的变化情况。
64.图21为实施例3中测试的aβ诱导cl2355秀丽线虫在各检测物作用下感知食物速率的变化情况。
65.图22为实施例3中在各检测物的影响下线虫cl2331菌株中的aβ沉积物的荧光图。
66.图23为实施例3中ppca、da和ta与aβ(1
‑
42)活性位点残基的三维相互作用和对接2d模型。
67.图24为实施例3中ta浓度从12.5μm增加到400μm的动力学结合感觉图。
68.图25为实施例3中ta的uhplc
‑
dad
‑
q/tof
‑
ms/ms分析谱图。
具体实施方式
69.下面结合附图,对本发明作详细的说明。
70.为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅用以解释本发明,并不用于限定本发明。
71.以下实施例中的化学品和试剂来源:
[0072]3‑
(4,5
‑
二甲基
‑2‑
噻唑基)
‑
2,5
‑
二苯基
‑2‑
h
‑
四唑溴化物(mtt,m2128)和硫黄素t(tht,596200)购自sigma(美国密苏里州圣路易斯)。
[0073]
milli
‑
q水是通过milli
‑
q整体净水系统(millipore,美国马萨诸塞州比勒里卡)制备的。
[0074]
乙腈购自anaqua chemicals supply(美国德克萨斯州休斯顿)。
[0075]
aβ(1
‑
42)从china peptides co.,ltd.(中国上海)获得。
[0076]
ez
‑
linknhslc
‑
lc
‑
生物素购自赛默飞世尔科技公司(美国马萨诸塞州沃尔瑟姆)。
[0077]
链霉亲和素(sa)生物传感器购自pall life sciences(美国纽约州华盛顿港)的fort
é
bio。
[0078]
hoechst 33342(b2261)和碘化丙锭(pi,p4170)购自sigma(美国密苏里州圣路易斯)。
[0079]
annexin v
‑
fitc/pe细胞凋亡检测试剂盒购自bd biosciences(美国加利福尼亚州圣何塞)。
[0080]
以下实施例中细胞培养主要是由以下方法得到的:
[0081]
pc
‑
12细胞购自美国菌种保藏中心(atcc;美国马里兰州罗克维尔)。
[0082]
pc
‑
12细胞在含有10%马血清、5%胎牛血清、50u/ml青霉素和50μg/ml链霉素的dmem培养基中培养。所有的细胞都被维持在37℃、5%的二氧化碳加湿培养箱中。
[0083]
mtt检测过程
[0084]
将100μl的pc
‑
12细胞种到96孔板中,密度为30,000个细胞/毫升。第二天,在没有或有aβ(1
‑
42)的情况下,用试验药物处理pc
‑
12细胞24小时。处理后,在孔中加入10μl的mtt溶液(5mg/ml),然后在37℃下孵育4小时。去除培养基,加入100μl dmso以溶解蓝色的结晶。用酶标仪(biotek,vt lab,美国)在570nm波长下测量溶液的光密度(od)值。细胞存活率根据以下公式计算。细胞活力(%)=100%
×
[od值(实验)
‑
od值(空白)]/[od值(对照)
‑
od值(空白)]。
[0085]
tht荧光检测
[0086]
在有或没有试验药物(包括ppac、dta和ta、姜黄素、茯苓或开心散提取物)的情况下,用pbs稀释2μl的aβ(1
‑
42)(1mm),最终体积设定为100μl。在0小时、24小时和48小时的孵化时间点,用pbs(ph=7.4)稀释50微升tht(20微升),并与100微升上述溶液混合,加入黑色96孔板中。然后,用酶标仪(biotek,vt lab,美国)读取溶液的od值,激发波长为450nm,发射波长为490nm。
[0087]
流式细胞仪分析
[0088]
用aβ(1
‑
42)在没有或有测试药物的情况下对种在6孔板中的pc
‑
12细胞进行24小时的处理,处理后的细胞被胰酶化并以2000rpm的速度离心。将细胞重新悬浮在250微升的1x缓冲液中,用2微升的碘化丙啶(pi)和1微升的异硫氰酸荧光剂(fitc)试剂(bd biosciences,san jose,美国加利福尼亚)在黑暗中进行15分钟染色。然后用facscalibur流式细胞仪(bd biosciences,san jose,美国加利福尼亚)对细胞进行分析。数据采集和分析由flowjo 7.6.1软件(tree star,san carlos,美国加利福尼亚)进行。
[0089]
hochest 33342/pi染色
[0090]
pc
‑
12细胞的死亡是通过hochest 33342/pi染色法测量的。简而言之,在没有或有aβ(1
‑
42)的情况下,用试验药物处理种在6孔板中盖玻片上的pc
‑
12细胞24小时。处理后,用新鲜的4%多聚甲醛(pfa)溶液固定15分钟,用pbs洗涤3次。然后,用5毫克/升的hoechst33342和5毫克/升的pi溶液对细胞进行染色5分钟。孵化后,用imagexpress micro4(molecular devices,美国)捕捉并合并显示同一区域内具有蓝色荧光的细胞核和显示红色荧光的细胞的代表性图像。然后,通过计算具有pi信号的细胞与具有蓝色信号的细胞的百分比来测量pc
‑
12细胞的死亡。
[0091]
线虫的株系和维持条件
[0092]
线虫株包括n2(wt);cl4176,dvis27[myo
‑
3p::a
‑
beta(1
‑
42)::let
‑
8513'utr)+rol
‑
6(su1006)]x;cl2122,dvis15[(ppd30.38)unc
‑
54(vector)+(pcl26)mtl
‑
2::gfp];cl2355,dvis50[pcl45(snb
‑
1::abeta 1
‑
42::3'utr(long)+mtl
‑
2::gfp]i;and cl2331,dvis37[myo
‑
3p::gfp::a
‑
beta(3
‑
42)+rol
‑
6(su1006)]均从秀丽隐杆线虫遗传学中心(cgc)得到。除非另有说明,所有的蠕虫都在线虫生长培养基(ngm)板上维持,并在20℃的培养箱中用大肠杆菌(e.coli)op50喂养。
[0093]
瘫痪试验
[0094]
通过测量ad秀丽隐杆线虫cl4176的瘫痪率,来测试药物在体内是否改善aβ的沉积。简而言之,将含有在肌肉细胞中表达的热诱导型人aβ(1
‑
42)转基因的cl4176线虫转移至ngm并用受试药物进行处理。将培养箱的温度从15℃改变为25℃以刺激aβ表达和聚集。在25℃下暴露36小时后,对线虫的瘫痪数量进行计数,最后统计瘫痪率。线虫在接触时无法移动其身体或在喂食时表现出清除细菌的“光晕”表现为瘫痪。
[0095]
线虫的行为在接触时无法移动其身体或在喂食时表现出清除细菌的“光晕”表现为瘫痪。
[0096]
食物感知测定
[0097]
用表达人aβ(1
‑
42)的cl2355菌株及其载体对照cl2122菌株来研究试验药物对食物感应行为的改善作用。简而言之,用大肠杆菌op50铺在一个内径约1厘米、外径约8厘米的环形板上。用ppac、dta、ta或茯苓提取物处理48小时后,用m9缓冲液清洗蠕虫,并将其放入有或没有大肠杆菌op50的ngm琼脂板中心。5分钟后,计数蠕虫的身体弯曲次数,时间为20s。根据以下公式计算身体弯曲率的减慢。减速率=(无食物的n
‑
有食物的n)/无食物的n。n代表线虫身体弯曲的总次数。
[0098]
aβ(3
‑
42)的聚集分析
[0099]
通过使用cl2331菌株研究aβ(3
‑
42)的聚集,该菌株在温度敏感的情况下表达体壁肌肉中与人aβ(3
‑
42)结合的gfp。简而言之,将经ppac、dta、ta或茯苓提取物处理的线虫进行孵育在23℃下诱导aβ(3
‑
42)聚集直至成虫第二天。处理后,将线虫固定在含有0.1%nan3的载玻片上。通过荧光显微镜(leica dm6b,leica microsystems gmbh,德国)捕获蠕虫的代表性图像。然后计算前区aβ沉积物的总数。
[0100]
分子对接
[0101]
为了进一步探索ppac、dta和ta与aβ的相互作用机制和结合模式,我们使用sybylx 2.0分子建模程序进行了分子对接研究。aβ(1
‑
42)的晶体结构(id:1fiq)是从结构生物信息学研究合作组织(rcsb)蛋白质数据库(pdb)中检索的。配体的sdf文件,包括ppac(cid:9805290)、dta(cid:15225964)和ta(cid:12314446)都是从ncbi pubchem数据库中获得。surflax
‑
dock程序被用来研究aβ和ppac、dta、ta之间的潜在相互作用模式。对接构象的可视化是由chimera分子图形软件和ligplot软件完成的。
[0102]
统计分析
[0103]
以下实施例所有数据均来自三个或更多独立实验,并以均值
±
标准差(sd)表示。使用graphpad prism 8.0统计软件(美国加利福尼亚州圣迭戈),采用单因素方差分析,然后进行tukey检验。p<0.5被认为在比较组之间具有统计学意义。
[0104]
实施例1
[0105]
黄芩中aβ纤维化抑制剂的验证和筛选
[0106]
yu l,wu a g,wong k w,et al.the new application ofuhplc
‑
dad
‑
tof/ms in identification ofinhibitors onβ
‑
amyloid fibrillation from scutellaria baicalensis[j].other,2019,10。该期刊中通过将将黄芩提取物预孵育aβ(1
‑
42),然后分析uhplc
‑
dad
‑
q/tof
‑
ms/ms,筛选了黄芩中aβ纤维化的潜在抑制剂。与单独使用黄芩提取物相比,在含有aβ的培养液中发现黄苷和黄苷素显着降低,表明黄苷和黄苷素是黄芩中有效的aβ纤维化抑制剂。
[0107]
实施例1采用本发明提供的高通量筛选方法进行验证
[0108]
制备黄芩提取物
[0109]
将黄芩进行称量、粉碎,然后用10倍体积的水进行蒸馏提取1h,重复提取3次,合并提取液,过滤,浓缩,然后将浓缩后的提取物用dmso(二甲基亚砜)溶解为400mg/ml的溶液,得到待测中草药提取物。
[0110]
制备生物素化的aβ(1
‑
42)肽
[0111]
5毫克的aβ(1
‑
42)肽被溶解在六氟异丙醇(hfip,sigma)溶液中,然后将hfip溶液分装到新的1.5ml试管中并在氮气流下干燥,然后,在试管中加入10μl的dmso(二甲基亚砜)和适当体积的pbs(包括15%的dmso和0.02%的tween 20),以获得100mg/ml的aβ(1
‑
42)的工作浓度。
[0112]
将摩尔比为5:1的生物素(ez
‑
linknhs
‑
lc
‑
lc
‑
biotin(thermo scientific,美国))与aβ(1
‑
42)肽溶液进行混合,然后再室温下孵育30min,得到生物素化的aβ(1
‑
42)肽。
[0113]
高通量筛选
[0114]
生物膜干涉分析
[0115]
用pbs(包括15%的dmso和0.02%的tween 20)将黄芩提取物进行稀释得到25、50、100、200、400μg/ml不同浓度的工作液。
[0116]
工作液和生物素化的aβ(1
‑
42)肽注入生物传感器中,进行结合
‑
解离,得到结合和解离图即图1所示,黄芩的缔合/解离结合曲线表明生物传感器层的光学厚度(nm)呈剂量依赖性增加,这表明黄芩提取物与aβ直接结合。表1中显示了由forte
ì
bio数据分析软件计算出的动力学常数,包括结合亲和力(k d),缔合速率常数(kon)和解离速率常数,分别为199μg/ml,6.18
×
10
+2
1/ms和1.23
×
10
‑11/s,表明黄芩提取物与aβ存在直接和可逆的相互作用。
[0117]
表1
[0118]
tcmkd(μg/ml)kon(1/ms)kdis(1/s)sb extract1996.18
×
10
+2
1.23
×
10
‑1[0119]
液质联用分析
[0120]
将400μg/ml黄芩提取物的工作液与生物素化的aβ(1
‑
42)肽在生物传感器中连续进行5次结合
‑
解离操作,收集解离液,同时,平行制备用于等体积400μg/ml黄芩提取物工作液作为对照液,将10μl解离液和对照液注入uhplc
‑
dad
‑
q/tof
‑
ms/ms仪器中进行分析,得到代表性总离子色谱图即图2所示,图2中,s1:不含sb提取物的解离缓冲液,s2:含sb提取物的解离缓冲液,s3:用于bli分析的sb提取物。与单独的黄芩提取物相比,在离解缓冲液中检测到了一些化合物,黄芩提取物中的保留时间,准确度,分子量,化学名称和所检测组分的配
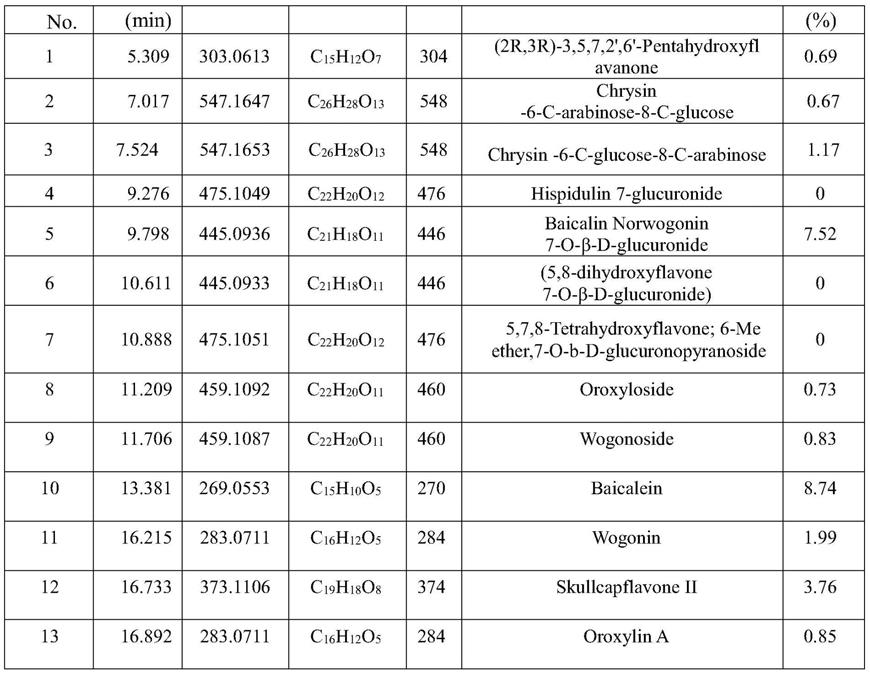
方列于表2中。其中,图3中显示黄芩中的黄苷和黄苷素的rba最高。因此,由实施例1采用本发明提供的高通量筛选方法验证了黄苷和黄苷素是黄芩中有效的aβ纤维化抑制剂的结论。
[0121]
表2
[0122][0123][0124]
实施例2
[0125]
制备开心散提取物
[0126]
将开心散(开心散中含有人参、远志、石菖蒲、茯苓)进行称量、粉碎,然后用10倍体积的水进行蒸馏提取1.5h,重复提取4次,合并提取液,过滤,浓缩,然后将浓缩后的提取物用dmso(二甲基亚砜)溶解为400mg/ml的溶液,得到待测中草药提取物。
[0127]
制备生物素化的aβ(1
‑
42)肽
[0128]
5毫克的aβ(1
‑
42)肽被溶解在六氟异丙醇(hfip,sigma)溶液中,然后将hfip溶液分装到新的1.5ml试管中并在氮气流下干燥,然后,在试管中加入10μl的dmso(二甲基亚砜)和适当体积的pbs(包括15%的dmso和0.02%的tween 20),以获得100mg/ml的aβ(1
‑
42)的工作浓度。
[0129]
将摩尔比为5:1的生物素(ez
‑
linknhs
‑
lc
‑
lc
‑
biotin(thermo scientific,美国))与aβ(1
‑
42)肽溶液进行混合,然后再室温下孵育30min,得到生物素化的aβ(1
‑
42)肽。
[0130]
高通量筛选
[0131]
生物膜干涉分析
[0132]
用pbs(包括15%的dmso和0.02%的tween 20)将开心散提取物进行稀释得到125、250、500、1000、2000、4000μg/ml不同浓度的工作液。
[0133]
工作液和生物素化的aβ(1
‑
42)肽注入生物传感器中,进行结合
‑
解离,得到结合和解离图即图4所示,开心散提取物的缔合/解离结合曲线表明生物传感器层的光学厚度(nm)呈剂量依赖性增加,这表明开心散提取物与aβ直接结合。表3中显示了由forte
ì
bio数据分析软件计算出的动力学常数,包括结合亲和力(k d),缔合速率常数(kon)和解离速率常数,分别为170μg/ml,1.15
×
102l/ms和2.6
×
10
‑21/s,表明开心散提取物与aβ存在直接和可逆的相互作用。
[0134]
表3
[0135]
tcmkd(μg/ml)kon(1/ms)kdis(1/s)kxs extract1701.15
×
10
+2
2.6
×
10
‑2[0136]
液质联用分析
[0137]
将4000μg/ml开心散提取物的工作液与生物素化的aβ(1
‑
42)肽在生物传感器中连续进行5次结合
‑
解离操作,收集解离液,同时,平行制备用于等体积4000μg/ml开心散提取物工作液作为对照液,将10μl解离液和对照液注入uhplc
‑
dad
‑
q/tof
‑
ms/ms仪器中进行分析,得到代表性总离子色谱图即图5所示,其中,s1:不含开心散提取物的解离缓冲液,s2:含有开心散提取物的解离缓冲液,s3:用于bli分析的开心散提取物,与单独的开心散提取物相比,在离解缓冲液中检测到了一些化合物,开心散提取物中的保留时间,准确度,分子量,化学名称和所检测组分的配方列于表4中。
[0138]
从图6至8中可以看出,在结合的化学成分中,其中有三种化合物的rba呈断层式高度,包括猪苓酸c(ppac),去氢土莫酸(dta)和土莫酸(ta),显示出与aβ的结合力最强。用开心散提取物的溶液。
[0139]
由上述高通量筛选方法,筛选出三萜酸类化合物为猪苓酸c、去氢土莫酸和土莫酸中的至少一种是aβ原纤维化抑制剂。
[0140]
表4
[0141]
[0142]
[0143]
[0144][0145]
实施例3
[0146]
从茯苓中分离纯化ppac,dta和ta
[0147]
首先,取茯苓粉,用乙酸乙酯试剂浸泡,然后用旋转蒸发仪旋转收集,重复3次,得到茯苓提取物。将茯苓乙酸乙酯提取物,使用硅胶层析柱进行粗分离,接取100ml馏分为一组,然后用质谱进行检测目标化合物,最后再使用超快速高效液相进行纯化。最后,成功分离出三种化合物,包括猪苓酸c、去氢土莫酸和土莫酸。
[0148]
图9中的代表性总离子色谱图显示了ppac,dta,ta和茯苓提取物的uhplc
‑
dad
‑
q/tof
‑
ms/ms分析。其中,s1:茯苓提取物;s2:ppac;s3:dta;s4:ta。从图10中显示,测得的ppac,dta和ta的准确质量分别为[mh]
‑
481.3388,[mh]
‑
483.3510和[mh]
‑
485.3368,并且它们的分子结构如图11所示,这与报道的化合物是一致的。
[0149]
tht荧光检测
[0150]
通过tht法研究了ppac,dta和ta对aβ(1
‑
42)原纤维形成的抑制作用。同时,用茯苓、开心散以及黄芩的提取物进行比较。tht与aβ(1
‑
42)的结合可以在482nm的波长处产生强烈的荧光信号,反映出aβ原纤维的形成。
[0151]
在0h,24h和48h的时间点,用分光光度计检测溶液的荧光强度。如图12至14所示,aβ(1
‑
42)显着增加了tht荧光,而ppac,dta,ta,茯苓、开心散以及姜黄素的提取物的处理可以降低荧光强度。其中,在这些测试药物中,ta和姜黄素提取物具有最佳的抑制作用。
[0152]
pc
‑
12细胞死亡实验
[0153]
越来越多的证据表明细胞外aβ原纤维逐渐积累并诱导细胞死亡的神经元。在目前
的实验中,我们研究了ppac,dta和ta对aβ(1
‑
42)诱导的pc
‑
12细胞的生存能力的保护作用。
[0154]
首先,通过mtt测定ppac,dta,ta,茯苓、开心散以及姜黄素的提取物在pc
‑
12细胞中的细胞毒性,结果如图15所示。
[0155]
然后,aβ(1
‑
42)诱导的pc
‑
12细胞经ppac(0.5μm)、dta(0.5μm)、ta(0.5μm)、茯苓(20μg/ml)、开心散(20μg/ml)和姜黄素(0.5μm)处理48h后,用四甲基偶氮唑盐比色法测定细胞活力,研究了它们对细胞活力的改善作用,结果如图16所示,ppac,dta,ta,茯苓、开心散以及黄芩的提取物显着提高了pc
‑
12细胞的活力。
[0156]
此外,aβ(1
‑
42)诱导的pc
‑
12细胞经ppac(0.5μm)、dta(0.5μm)、ta(0.5μm)或茯苓(20μg/ml)处理48h后,收集细胞用annexin v
‑
fitc/pe凋亡试剂盒进行流式细胞仪分析,我们发现茯苓提取物,ppac,dta和ta显着降低了经aβ(1
‑
42)处理的pc
‑
12细胞的细胞死亡,测试结果如图17所示。
[0157]
此外,aβ(1
‑
42)诱导的pc
‑
12细胞经ppac(0.5μm)、dta(0.5μm)、ta(0.5μm)或茯苓(20μg/ml)处理48h后,用hoechst33324/pi染色5min。采集同一视野内具有hoechst或pi信号的细胞的代表性图像,并进行融合(放大倍率,
×
10)。比例尺:100μm。柱状图显示pc
‑
12细胞的pi/hoechst信号的比率,hoechst/pi染色结果表明茯苓提取物,ppac,dta和ta可以抑制细胞死亡,这是由带有pi信号的细胞数量减少所揭示的,测试结果如图18所示。
[0158]
综上,以上数据表明ppac,dta和ta抑制aβ(1
‑
42)诱导的pc
‑
12细胞的死亡。
[0159]
秀丽隐杆线虫的行为能力测试
[0160]
为了研究ppac,dta和ta在ad体内模型中的神经保护作用,使用了秀丽隐杆线虫(一种广泛用于神经变性疾病的模型生物)。
[0161]
测试结果如图19所示,将aβ(1
‑
42)诱导的cl4176线虫分别用茯苓提取物,ppac,dta或者ta在一定浓度下处理72h后,在显微镜下(放大倍数:10x)拍摄有代表性的线虫图像。比例尺:100μm。柱状图显示未瘫痪的线虫(n>60)数量的百分率。另外,aβ诱导了秀丽线虫cl2355及其对照菌株cl2122用于研究食物的搜寻行为,结果如图20所示,将aβ诱导的线虫cl2355和对照菌株cl2122分别用茯苓提取物、ppac、dta和ta在一定浓度下处理72h,然后在无食物或有食物的情况下,通过计数每20s在ngm板上的线虫弯曲数量来评价其食物感知行为。柱状图显示线虫感知食物后的速度减缓率。在cl2122菌株的对照组和处理组之间没有观察到显著性。如图21所示,茯苓提取物,ppac,dta和ta的处理可以显著提高cl2355菌株的减慢速度。综上,以上数据表明ppac,dta或ta改进了秀丽隐杆线虫模型的行为功能。
[0162]
秀丽隐杆线虫中aβ沉积物变化测试
[0163]
除了行为能力外,我们还测量了线虫cl2331菌株中的aβ沉积物,该菌株在体壁肌肉细胞中温度敏感地表达了与gfp缀合的人aβ(1
‑
42),测试结果如图22所示,用茯苓提取物、ppac、dta、ta在一定浓度下处理线虫cl233172h,用荧光显微镜(放大倍率:40x)拍摄线虫头部有代表性的图像。柱状图显示了线虫cl2331前部的aβ沉积物总数;bars,s.d.,***p≤0.001,n=20。结果显示,茯苓提取物,ppac,dta和ta的总数显着减少了aβ沉积在前区,这表明ppac,dta和ta抑制了体内aβ沉积的形成。
[0164]
与aβ结合(1
‑
42)实验测试
[0165]
为了找到更理想的ppac,dta和ta在aβ蛋白的结合位点并确定其理论结合模式,进行了基于分子对接的计算。如图23所示,ppca、da和ta与aβ(1
‑
42)形成的复合物的结合姿
态。顶部:配体结合口袋表面的3d相互作用形状和极性,底部:交互组件的停靠2d模型详细信息。ppca、da和ta以棒状显示,关键残基以棒状突出显示。虚线代表氢键。证明了这些化合物与aβ蛋白的疏水口袋相互作用。例如图中显示,ppac与asp a:7和his a:14残基形成氢键,并与诸如phe a:19,glu a:22,glna a:15和glu a:11等残基形成范德华力。dta与glua:11,asna:27和hisa:14残基形成氢键,并与残基如asp a:7,gln a:15,glu a:22形成范德华力。与glu a:11,asn a:27,asp a:23残基形成氢键,并与残基如asp a:23,gln a:15,glu a:22和phe a:19形成范德华力。此外,ppac,dta和ta的结合分数分别为4.06、4.76和5.05。
[0166]
因此,ppac,dta和ta具有与aβ的强结合特性。基于上述体外和体内结果,我们进一步使用bli技术验证了ta对aβ(1
‑
42)的结合。如图24所示,响应/结合(nm)表示ta与aλ(1
‑
42)相互作用时光谱位移(β)所反映的传感器层的光学厚度。当缔合速率等于解离时的稳态响应,即由平坦曲线表示的平衡结合信号(req)。ta表现出与aβ(1
‑
42)的直接和可逆相互作用,如传感器层光学厚度(nm)的剂量依赖性增加所揭示。另外,表5显示了动力学常数,包括结合亲和力(k d),缔合速率常数(kon)和解离速率,分别为49.9μm,1.49
×
10
+2
1/ms和7.42
×
10
‑31/s。此外,如图25中显示,s1代表ta,s2代表解离缓冲液,s3代表空白缓冲液。uhplc
‑
dad
‑
q/tof
‑
ms/ms结果表明,可以在解离的溶液中检测到ta。
[0167]
总的来说,ta是开心散提取物中筛选出的化合物中与aβ的结合亲和力最高的化合物。
[0168]
实施例3中,通过体外和体内生物测定表明,dta,ta和ppac可以抑制aβ纤维的形成,降低pc
‑
12细胞中aβ的细胞毒性,并改善行为秀丽隐杆线虫的能力。将来,这种方法也将用于筛查靶向其他病理蛋白(例如tau,α
‑
突触核蛋白和与神经退行性疾病相关的亨廷顿蛋白(htt))的中药潜在抑制剂。
[0169]
表5
[0170]
compoundkd(μm)kon(1/ms)kdis(1/s)ta49.91.49
×
10
+2
7.42
×
10
‑3[0171]
本发明提供的筛选aβ纤维形成抑制剂的高通量方法,通过生物膜干涉分析和液质联用分析组合使用,最终根据代表性总离子色谱图分析即可筛选出aβ纤维形成抑制剂化合物。该筛选方法,操作简单,占用时间少,准确度高,精准性高,重复性好,便于广泛的推广应用,对从中药中轻松的筛选出aβ纤维形成抑制剂具有十分重要的意义,加速了未来抗ad药物的发现和发展进程。
[0172]
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1