一种夹心式在线衍生堆积富集方法及其在铁皮石斛中的应用
1.本发明涉及中药中氨基酸的分离富集领域,尤其是涉及一种夹心式在线衍生堆积富集方法。
背景技术:
2.毛细管电泳(ce)因其强大的分离功能被广泛应用于许多物质的分离和检测,如dna、酶、无机离子和复杂样品中的有机小分子。然而,样品注入体积小、光程短的缺点阻碍了毛细管电泳的发展。为了解决这些问题,人们建立了多种方法来提高ce的检测效率,其中在线富集技术中的堆积技术受到了广泛的关注。各种堆积方法,包括场放大样品堆积(fass)、场增强样品注入(fesi)、大体积样品堆积(lvss)和胶束
‑
溶剂堆积(mss)都致力于提高毛细管电泳中多种极性物质的检测灵敏度,例如专利cn110160856b公开了一种基于fass
‑
mcds在线富集测定生物碱含量的方法,分析时间短、无需用到有机溶剂,而且灵敏度得到了显著提高,富集倍数可达169~218倍,为复杂样品基质中生物碱的测定提供了新选择。但是在堆积过程中,与背景溶液(bgs)相比,溶解在电导率较低的溶液中的分析物在样品区的电泳迁移速度要快于背景溶液区,然后化合物堆积在背景溶液和样品之间的边界处,堆积的分析物被分离并检测到单个区域。因此,分析物通过堆积可获得更好的分离效率和检测灵敏度。近年来,联用技术是当前的技术热点,ce与质谱联用(ce
‑
ms)和ce与激光诱导荧光联用(ce
‑
lif)大大提高了检测灵敏度。但由于检测过程不稳定、仪器昂贵,在推广联合检测技术的方向上有一定的阻碍作用。紫外
‑
可见光检测器作为毛细管电泳中最通用的检测器,与其他方法相比是经济的,但是对于一些复杂基质中的分析物的检测,没有紫外吸收,需要开发新的技术来提高检测灵敏度和富集效率。
3.氨基酸作为人体内最重要的小分子之一,不仅是蛋白质的基本单位,而且对人类的生理调节具有重要意义。由于其在生命和健康中的重要作用,一直是学术界研究的重点,建立一种更快、更有效的检测方法是一个重要的研究方向。由于大多数氨基酸都没有发色团,因此用衍生剂与氨基酸发生衍生化反应对于增强光谱特性和提高检测效率至关重要。传统的柱前衍生后,反应物的检测信号强,但柱前衍生操作繁琐,试剂用量大。毛细管在线衍生技术因其简单、环保、高效等优点而受到广泛关注。在线衍生化技术将样品和衍生剂直接注入毛细管中,在电场作用下进行反应,弥补了氨基酸分析中的这些不利影响,使整个分析过程更加简单、高效。但与柱前衍生相比,在线衍生的信号强度明显降低。目前,ce已被应用于测定人体细胞和组织、中药和药用植物中的氨基酸。对于毛细管电泳在线衍生化中,有关氨基酸与铜离子的在线络合、微透析、电泳介导的微量分析等已有许多相关报道。这些创新技术对提高在线衍生的效率做出了一定的贡献,但其中有些方法的检测灵敏度还有待进一步提高,一些衍生剂与氨基酸反应需要有毒催化剂的参与。因此,进一步探索开发一种更安全高效的在线检测氨基酸的方法具有十分重要的意义。
技术实现要素:
4.本发明为了克服传统氨基酸检测方法无法实现多种氨基酸高效分离的问题,提供一种夹心式在线衍生堆积富集方法,利用毛细管电泳在线夹心衍生堆积法检测中药中的多种氨基酸,不仅实现了氨基酸的快速检测,也极大地提高了毛细管电泳样品在线富集的灵敏度。
5.为了实现上述目的,本发明采用以下技术方案:一种夹心式在线衍生堆积富集方法,包括以下步骤:(1)用纯净水和硼砂配制硼砂缓冲液,稀释后作为运行缓冲液和样品基质;(2)将氨基酸储备液用硼砂缓冲液稀释制备成分析用的标准对照品溶液;(3)将nbd
‑
cl溶于乙腈配置成衍生剂;(4)活化毛细管柱之后,先用运行缓冲液冲洗,再按照样品、衍生剂、样品的进样顺序依次注射分析物,在毛细管中进行衍生反应,反应结束后衍生产物在压力进样下进入毛细管中进行富集和分离检测。
6.作为优选,步骤(1)所述运行缓冲液的浓度为20
‑
50mm。作为进一步优选,步骤(1)所述运行缓冲液的浓度为40mm。
7.作为优选,步骤(1)所述样品基质的浓度为0
‑
50mm。作为进一步优选,步骤(1)所述样品基质的浓度为40mm。
8.作为优选,步骤(2)所述标准对照品溶液的浓度为0.1
‑
0.3mg/ml。
9.作为优选,步骤(3)所述衍生剂的浓度为30
‑
50mm。
10.作为优选,步骤(4)所述样品为氨基酸,其进样时间为40
‑
120s,进一步优选为80s。
11.作为优选,步骤(4)所述衍生剂进样时间为3
‑
12s,进一步优选为6s。
12.作为优选,步骤(4)所述衍生反应的时间为90
‑
360s,进一步优选为360s。衍生反应的时间也称等待时间,为样品注射后到分析程序开启前的一段化合物的反应时间。
13.作为优选,步骤(4)所述压力进样的压强为60
‑
100mbar。作为进一步优选,步骤(3)所述压力进样的压强为90mbar。
14.本发明还提供上述方法在铁皮石斛中的应用,用于铁皮石斛样品中氨基酸的在线衍生和堆积。铁皮石斛的营养成分丰富,富含多种氨基酸,对它含有的氨基酸进行定性和定量检测是其质量鉴别的必要条件。本发明建立了一种利用毛细管电泳在线夹心衍生和堆积的新方法,用于预富集中药样品中的多种氨基酸,并与柱前衍生法进行了比较,利用毛细管电泳进行夹心式的在线衍生更加方便、高效。将氨基酸、4
‑
氯
‑7‑
硝基
‑
1,2,3
‑
苯并恶二唑(nbd
‑
cl)、氨基酸依次注入毛细管中,几分钟的反应后进行分离检测。进一步优化了一系列实验参数,获得了最佳的分离效率和灵敏度提高,并应用于铁皮石斛中9种的氨基酸的分析。
15.因此,本发明的有益效果为:(1)提出了一种基于毛细管电泳的在线夹心衍生和堆积策略,并成功地应用于中药样品中多种氨基酸的预富集;(2)提出的毛细管电泳在线夹心式衍生和层叠方法可用于复杂样品中多种氨基酸的定量测定和快速分离;(3)本发明用于铁皮石斛样品中氨基酸的在线衍生和堆积。
附图说明
16.图1是在线夹心式衍生和堆叠过程的机理图。
17.图2是aas与nbd
‑
cl衍生化反应的反应原理图。
18.图3是实施例1
‑
4不同缓冲液浓度下的色谱图,其中a:20mm,b:30mm,c:40mm,d:50mm。氨基酸:l
‑
精氨酸(1),l
‑
赖氨酸(2),亮氨酸(3),缬氨酸(4),l
‑
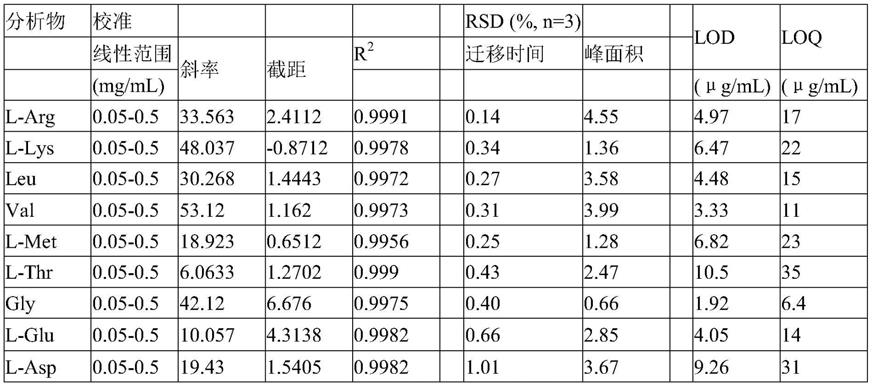
蛋氨酸(5),l
‑
苏氨酸(6),甘氨酸(7),nbd
‑
cl(8),l
‑
谷氨酸(9),l
‑
天冬氨酸(10)。样品基质浓度为30mm。ce条件:在反应缓冲液中连续注入0.2mg/ml aas,40mm nbd
‑
cl,并在反应缓冲液中以80mbar的压强在25℃下连续注入0.2mg/ml aas。
19.图4是实施例5
‑
9不同压强下(60mbar、70mbar、80mbar、90mbar、100mbar)氨基酸衍生物的峰面积图。ce条件:分离电压25kv;毛细管温度设置为25℃;进样时间分别为10s、6s、10s,等待时间为180s。
20.图5是实施例10
‑
13不同衍生剂注入时间下(a:3s,b:6s,c:9s,d:12s)的色谱图。其他条件与图4相同。
21.图6是实施例14
‑
18不同等待时间下(90s、180s、270s、300s、360s)氨基酸衍生物的峰面积图。其他条件与图5相同。
22.图7是实施例19
‑
23不同样品基质浓度下的色谱图,其中,a:氨基酸溶于水;b:氨基酸溶于20mm硼砂缓冲液中;c:氨基酸溶于30mm硼砂缓冲液中;d:氨基酸于40mm硼砂缓冲液中;e:氨基酸溶于50mm硼砂缓冲液中。ce条件:进样压强90mbar;分离电压25kv;毛细管温度设置为25℃;进样时间分别为10s、6s、10s,等待时间为360s。
23.图8是实施例24
‑
28不同样品注入时间下(40s、60s、80s、100s、120s)氨基酸衍生物的峰面积图。其他条件与图7相同。
24.图9是铁皮石斛的电泳图谱。其中,a:铁皮石斛电泳图谱。ce条件:在缓冲液中以25℃、90mbar的压强连续注入提取物,40mm nbd
‑
cl,提取物各30s、6s、30s;等待时间为180s。
具体实施方式
25.下面通过具体实施例,对本发明的技术方案做进一步说明。
26.本发明中,若非特指,所采用的原料和设备等均可从市场购得或是本领域常用的,实施例中的方法,如无特别说明,均为本领域的常规方法。
27.一、测试条件优化1、实施例1
‑
4研究缓冲液浓度对检测效果的影响缓冲液浓度有利于分析物的预富集,不仅控制了电泳介质的离子强度,而且对电导也有很大的影响。因此,实施例1
‑
4分别研究了缓冲液浓度为20、30、40、50mm的影响,并通过溶解硼砂得到缓冲液。衍生的其他实验条件是一致的,包括在反应缓冲液中以80mbar的压强在25℃下连续注入0.2mg/ml氨基酸,40mm nbd
‑
cl,0.2mg/ml氨基酸10s,6s,10s,等待3分钟后开始富集分析。
28.具体的ce条件为如下检测波长:475nm;柱温:25℃。
29.毛细管柱:内径50μm,外径375μm,长度48.5cm,有效长度40cm。在第一次使用之前,新的毛细管柱要用1.0m naoh溶液冲洗30分钟,0.1m naoh溶液冲洗15分钟,纯水冲洗10分
钟,运行缓冲液冲洗30分钟。每天使用前用1.0m naoh溶液冲洗10分钟,0.1m naoh溶液冲洗10分钟,纯水冲洗5分钟,运行缓冲液冲洗5分钟。为了实现良好的重复性,两针间用0.1m naoh溶液冲洗2分钟,纯水冲洗2分钟,运行缓冲液冲洗2分钟。
30.分离电压:25kv。
31.缓冲体系:运行缓冲液由20
‑
50mm硼砂缓冲液组成(ph=9.3)。
32.数据记录:hp化学工作站(agilent)。
33.图3显示,50mm的硼砂浓度使九种分析物有了最佳峰高和分辨率。随着硼砂缓冲液浓度的增加,堆积效果增强,样品容量增大,分析灵敏度和峰高提高。观察到,由于慢电渗流(eof)和表观迁移率的直接影响,较高浓度的硼砂逐渐延长分析物的迁移时间(6.605至8.804min)。其原因是缓慢的eof有利于氨基酸衍生物在毛细管中的分布平衡,有利于堆积过程。50mm硼砂缓冲液有利于缬氨酸和l
‑
蛋氨酸的分离(3.35),20mm硼砂缓冲液的分离度为1.20。当浓度达到50mm时,l
‑
谷氨酸和l
‑
天冬氨酸的峰不能很好地分离,分离度为1.10。同时,随着缓冲液浓度的增加,电流显著增加(43.5~83.9μa)。硼砂缓冲液离子强度的增加导致电导的增加,而电导会产生大量的焦耳加热,系统温度过高会导致峰宽、峰高增加,分辨率降低。由于堆积过程的影响,峰宽没有明显增加。虽然迁移时间在40mm和50mm时相似,但在50mm(1.10)时l
‑
glu和l
‑
asp的分辨率低于40mm(8.94)。另外,硼砂缓冲液浓度越高,产生的焦耳热越大。因此,选择40mm硼砂缓冲液浓度进行进一步的研究。
34.2、实施例5
‑
9研究压强对检测效果的影响将样品注入毛细管电泳柱有两种常见的方式:流体动力注入和电动注入。在流体动力注射中,通过在毛细管两端施加压力差,一小部分样品被迫进入毛细管。压力在在线预富集技术中起着重要的作用,它影响进样量、检测灵敏度和叠加效果。本发明实施例5
‑
9分别考察了60、70、80、90、100mbar压强下对检测效果的影响,运行缓冲液浓度为40mm的硼砂,在反应缓冲液中连续注入0.2mg/ml氨基酸,40mm nbd
‑
cl,0.2mg/ml氨基酸10s,6s,10s,等待3分钟后开始富集分析。
35.具体ce条件如下检测波长:475nm;柱温:25℃。
36.毛细管柱:内径50μm,外径375μm,长度48.5cm,有效长度40cm。在第一次使用之前,新的毛细管柱要用1.0m naoh溶液冲洗30分钟,0.1m naoh溶液冲洗15分钟,纯水冲洗10分钟,运行缓冲液冲洗30分钟。每天使用前用1.0m naoh溶液冲洗10分钟,0.1m naoh溶液冲洗10分钟,纯水冲洗5分钟,运行缓冲液冲洗5分钟。为了实现良好的重复性,两针间用0.1m naoh溶液冲洗2分钟,纯水冲洗2分钟,运行缓冲液冲洗2分钟。
37.分离电压:25kv。
38.缓冲体系:运行缓冲液由40mm硼砂缓冲液组成(ph=9.3)。
39.数据记录:hp化学工作站(agilent)。
40.图4显示氨基酸衍生物的峰面积随压强的增加而增加,特别是当压强在80mbar到90mbar之间变化时。当压强增加到100mbar时,峰面积增加缓慢,l
‑
met和l
‑
asp衍生物的峰面积减少。这种现象可以解释为堆积技术降低了分析物谱带的宽度,因此,尽管注入体积比通常的注入体积大,但分辨率或效率没有下降。然而,过量的进样会导致堆积失败,因为在分析物到达检测器之前,程序无法完成。为了获得更好的堆积效率,确定了最佳压强为
90mbar。
41.3、实施例10
‑
13研究衍生剂进样时间对检测效果的影响在线衍生中,衍生剂与氨基酸直接在毛细管中反应,并检测产物。为了获得更好的预富集和在线衍生化效率,实施例10
‑
13分别研究了衍生剂的进样时间为3、6、9、12s时对检测结果的影响。选取40mm的nbd
‑
cl作为衍生剂,以90mbar压强分别注入0.2mg/ml氨基酸、40mm nbd
‑
cl、0.2mg/ml氨基酸溶液10s、(3、6、9、12)s、10s,等待3分钟后开始富集分析。
42.具体ce条件如下检测波长:475nm;柱温:25℃。
43.毛细管柱:内径50μm,外径375μm,长度48.5cm,有效长度40cm。在第一次使用之前,新的毛细管柱要用1.0m naoh溶液冲洗30分钟,0.1m naoh溶液冲洗15分钟,纯水冲洗10分钟,运行缓冲液冲洗30分钟。每天使用前用1.0m naoh溶液冲洗10分钟,0.1m naoh溶液冲洗10分钟,纯水冲洗5分钟,运行缓冲液冲洗5分钟。为了实现良好的重复性,两针间用0.1m naoh溶液冲洗2分钟,纯水冲洗2分钟,运行缓冲液冲洗2分钟。
44.分离电压:25kv。
45.缓冲体系:运行缓冲液由40mm硼砂缓冲液组成(ph=9.3)。
46.数据记录:hp化学工作站(agilent)。
47.图5色谱图显示目标化合物的灵敏度和峰面积在6s比3s好,但在6s到12s的范围内也观察到较差的分离效率和糟糕的基线。由此可以解释衍生剂的进样时间对进样量的影响。nbd
‑
cl作为反应物的用量影响反应速率和荧光衍生物的产率。当nbd
‑
cl用量小于氨基酸用量时,随着衍生化剂用量的增加,产品收率增加。因此,在3s~6s范围内获得了较高的检测灵敏度。但随着进样时间的延长,在线预富集技术出现了明显的增宽峰,导致分离效率下降,达不到基线分离。此外,考虑到nbd
‑
cl在乙腈中的溶解性,随着nbd
‑
cl进样量的增加,毛细管中乙腈的引入量增加。在毛细管电泳中,一定量的有机相会抑制eof,导致运行电流消失。因此,选择6s作为注入时间。
48.4、实施例14
‑
18研究衍生反应时间对检测效果的影响实施例14
‑
18分别研究了衍生反应时间90s、180s、270s、300s、360s对反应程度和氨基酸灵敏度的影响。衍生反应时间也称等待时间,为样品注射后到分析程序开启前的一段化合物的反应时间,以90mbar压强分别注入0.2mg/ml氨基酸、40mm nbd
‑
cl、0.2mg/ml氨基酸溶液10s、6s、10s,等待相应时间后开始富集分析。
49.具体ce条件如下检测波长:475nm;柱温:25℃。
50.毛细管柱:内径50μm,外径375μm,长度48.5cm,有效长度40cm。在第一次使用之前,新的毛细管柱要用1.0m naoh溶液冲洗30分钟,0.1m naoh溶液冲洗15分钟,纯水冲洗10分钟,运行缓冲液冲洗30分钟。每天使用前用1.0m naoh溶液冲洗10分钟,0.1m naoh溶液冲洗10分钟,纯水冲洗5分钟,运行缓冲液冲洗5分钟。为了实现良好的重复性,两针间用0.1m naoh溶液冲洗2分钟,纯水冲洗2分钟,运行缓冲液冲洗2分钟。
51.分离电压:25kv。
52.缓冲体系:运行缓冲液由40mm硼砂缓冲液组成(ph=9.3)。
53.数据记录:hp化学工作站(agilent)。
54.结果如图6所示。结果表明,随着时间的推移,峰面积有增加的趋势,在90s时没有峰检测到。但当等待时间从270s变为300s时,部分分析物的峰面积减小,360s时l
‑
谷氨酸和l
‑
天冬氨酸的峰面积甚至低于270s;随着时间从300s到360s的变化,所有氨基酸的峰面积都在不断增加。其原因是:毛细管电泳在线压力进样衍生化使得试剂和氨基酸在毛细管内混合,衍生物在毛细管内分离。在电场作用下,随着等待时间的延长,样品的反应更加充分,出现了增敏现象。在360s的等待时间下,峰值面积有了明显的改善。因此,最终选择360s作为等待时间,以减少时间消耗。
55.5、实施例19
‑
23研究样品基质浓度对检测效果的影响由于样品基质浓度对富集性能有很大影响,实施例19
‑
23分别对样品基质浓度进行了研究,其中,实施例19将氨基酸溶于水,实施例20将氨基酸溶于20mm硼砂缓冲液中,实施例21将氨基酸溶于30mm硼砂缓冲液中,实施例22将氨基酸于40mm硼砂缓冲液中,实施例23将氨基酸溶于50mm硼砂缓冲液中。所选运行缓冲液的浓度为40mm的硼砂缓冲溶液。以90mbar压强分别注入0.2mg/ml氨基酸、40mm nbd
‑
cl、0.2mg/ml氨基酸溶液10s、6s、10s,等待360s后开始富集分析。
56.具体ce条件如下检测波长:475nm;柱温:25℃。
57.毛细管柱:内径50μm,外径375μm,长度48.5cm,有效长度40cm。在第一次使用之前,新的毛细管柱要用1.0m naoh溶液冲洗30分钟,0.1m naoh溶液冲洗15分钟,纯水冲洗10分钟,运行缓冲液冲洗30分钟。每天使用前用1.0m naoh溶液冲洗10分钟,0.1m naoh溶液冲洗10分钟,纯水冲洗5分钟,运行缓冲液冲洗5分钟。为了实现良好的重复性,两针间用0.1m naoh溶液冲洗2分钟,纯水冲洗2分钟,运行缓冲液冲洗2分钟。
58.分离电压:25kv。
59.缓冲体系:运行缓冲液由40mm硼砂缓冲液组成(ph=9.3)。
60.数据记录:hp化学工作站(agilent)。
61.如图7所示,当氨基酸溶解在水中时,检测到的灵敏度高于溶解在20mm硼砂缓冲液中时的灵敏度。样品基质浓度从20mm增加到40mm时,峰面积和峰高得到改善,而迁移时间基本保持不变,但所有分析物的基线分离在50mm处不稳定。其主要原因为样品基质和缓冲液浓度的不同导致了它们的两种电导率之间的差异。如果样品区的导电率低于运行缓冲区,则由于电导率与电场呈反比关系,样品塞中的电场强度较高。因此,分析物迅速向样品/缓冲区边界移动,在那里它们的电泳迁移速度降低,形成分析物的“堆积”。由于有机相是通过夹心进样辅助在线衍生引入的,所以样品区电导低于运行缓冲液区电导。此外,电流从62.5变为64.3μa当底物浓度从0mm增加到30mm时,电流从64.3减小到61.8μa当浓度在30
‑
50mm之间变化时。根据以上结果,最终选择了40mm样品基质的浓度。
62.6、实施例24
‑
28研究样品进样时间对检测效果的影响以往的研究表明,进样时间的长短是影响分析物富集率和灵敏度的重要因素之一。为此,实施例24
‑
28在上述优化条件下分别对样品基质的不同进样时间(40s、60s、80s、100s、120s)进行了优化,而nbd
‑
cl的进样时间分别调整为6s、6s、10s、12s和12s。
63.具体ce条件如下:检测波长:475nm;柱温:25℃。
64.毛细管柱:内径50μm,外径375μm,长度48.5cm,有效长度40cm。在第一次使用之前,
新的毛细管柱要用1.0m naoh溶液冲洗30分钟,0.1m naoh溶液冲洗15分钟,纯水冲洗10分钟,运行缓冲液冲洗30分钟。每天使用前用1.0m naoh溶液冲洗10分钟,0.1m naoh溶液冲洗10分钟,纯水冲洗5分钟,运行缓冲液冲洗5分钟。为了实现良好的重复性,两针间用0.1m naoh溶液冲洗2分钟,纯水冲洗2分钟,运行缓冲液冲洗2分钟。
65.分离电压:25kv。
66.缓冲体系:运行缓冲液由40mm硼砂缓冲液组成(ph=9.3)。
67.数据记录:hp化学工作站(agilent)。
68.从图8可以看出,随着进样时间从40到80s的变化,峰面积增加,但是当样品进样时间增加到80s时,灵敏度不再增加,分析物的分辨率降低。当进样时间大于80s时,leu、val和l
‑
met的峰未完全分离,分辨率小于1.20。这说明氨基酸与nbd
‑
cl的体积比影响产品的收率;同时,在线预富集技术的目的是通过比常规进样量大的进样量来提高氨基酸的灵敏度。因此,随着进样时间的增加,检测灵敏度有所提高,但堆积技术使样品过载,样品区扩散,导致峰重叠。考虑到两种分析物的灵敏度,选择80s作为进样时间作为实验条件,进样顺序为0.2mg/ml氨基酸、40mm nbd
‑
cl、0.2mg/ml氨基酸溶液40s、10s、40s,等待360s。
69.7、重复性考察为了全面评价在线夹心衍生和叠加的方法性能,对线性范围、精密度(rsd%)、检出限(lod)、定量限(loq)和回收率进行了验证,并显示在表1中。标准分析溶液中含有浓度为0.05、0.1、0.2的每种氨基酸,取混合标准溶液中0.4和0.5mg/ml,建立校准曲线。在所研究的浓度范围内,获得了校正峰面积的良好线性值(r2>0.995)。在信噪比为3:1和10:1的条件下分别测定了lod和loq值,结果表明lod和loq值低至1.92μg/ml和6.4μg/ml。通过连续三次注射0.1mg/ml氨基酸标准混合物来研究重复性。迁移时间的相对标准偏差(rsd)为0.14~1.01%,峰面积小于4.55%,具有良好的精密度。通过一系列的方法学检验,证明了该方法的有效性。
70.表1.线性回归数据、lod和loq二、对铁皮石斛样品进行了游离氨基酸分析建立了在线夹心式衍生富集法,对铁皮石斛样品进行了游离氨基酸分析。典型的电泳图如图9所示。回收率试验是通过在每种氨基酸浓度为0.1mg/ml的标准混合氨基酸中加入样品(铁皮石斛)来设计的,如表2所示,检测到了7种氨基酸,但没有得到亮氨酸和左旋
苏氨酸的峰,测定的回收率在66.4%~102.6%之间。用标准加入法和氨基酸标准迁移时间法鉴定了衍生物的峰。结果表明,该方法可用于中药样品的氨基酸。
71.表2.加标样品的分析结果分析物 铁皮石斛回收率(%)
ꢀꢀ
(ng/g) l
‑
arg 41102.6l
‑
lys 10381.1leu
ꢀ‑
86.9val 2.166.4l
‑
met 2167.9l
‑
thr
ꢀ‑
69.5gly 8685.2l
‑
glu 14894.7l
‑
asp 20690.8以上所述,仅是本发明的较佳实施例而已,并非对本发明作任何形式上的限制,虽然本发明已以较佳实施例揭露如上,然而并非用以限定本发明,任何熟悉本专业的技术人员,在不脱离本发明技术方案范围内,当可利用上述揭示的技术内容作出些许更动或修饰为等同变化的等效实施例,但凡是未脱离本发明技术方案内容,依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与修饰,均仍属于本发明技术方案的范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1