一种富马酸丙酚替诺福韦制剂中丙酚替诺福韦的检测方法与流程
1.本发明属于药物分析技术领域,具体涉及一种富马酸丙酚替诺福韦制剂中丙酚替诺福韦的检测方法。
背景技术:
2.富马酸丙酚替诺福韦(taf)是gilead sciences inc.(吉利德)开发的口服核苷转录酶抑制剂。于2016年11月10日获得fda批准上市,商品名为vemlidy,上市规格为25mg;2018年11月8日在我国获批上市,商品名为“韦立得/vemlidy”,规格为25mg。参比制剂为黄色薄膜包衣的无刻痕速释(ir)片剂,该片剂须在没有任何改变的情况下直接吞服。因此,计划开发的仿制药产品也将为黄色薄膜包衣的无刻痕速释(ir)片剂。说明书中的每日最大给药剂量为25mg(即每次一片,一天一次)。每次一片,随餐服用。
3.丙酚替诺福韦是富马酸丙酚替诺福韦片中的主成分,化学名为9
‑
[(r)
‑2‑
[[(s)
‑
[[(s)
‑1‑
(异丙氧基羰基)乙基]氨基]苯氧基氧膦基]甲氧基]丙基]腺嘌呤,结构式如下所示:
[0004][0005]
含量的研究是药品研发的一项重要内容,它包括选择合适的分析方法,准确地测定主成分的含量,并给出合理限度。这一研究贯穿于药品研发的整个过程,所以规范地进行含量的研究,并将其控制在一个可控、合理的限度范围之内,将直接关系到上市药品的质量及安全性。目前,国内外测定富马酸丙酚替诺福韦制剂含量的药物分析方法的文献较少,没有该品种的标准收载,因此,开发一种操作简便、准确度好的富马酸丙酚替诺福韦制剂中丙酚替诺福韦的检测方法,对实现丙酚替诺福韦的质量控制具有重要意义。
技术实现要素:
[0006]
针对现有技术中存在的上述不足,本发明的目的在于提供一种富马酸丙酚替诺福韦制剂中丙酚替诺福韦的检测方法,所述检测方法操作简便、准确度好,能够提升对富马酸丙酚替诺福韦制剂的质量控制水平。
[0007]
为实现上述发明目的,本发明采用的技术方案如下:
[0008]
本发明提供了一种富马酸丙酚替诺福韦制剂中丙酚替诺福韦的检测方法,所述丙酚替诺福韦具有式ⅰ结构,
[0009][0010]
所述检测方法采用反相高效液相色谱法,包括如下步骤:
[0011]
制备富马酸丙酚替诺福韦供试品溶液;
[0012]
制备富马酸丙酚替诺福韦对照品溶液;
[0013]
将所述供试品溶液与所述对照品溶液进行高效液相色谱检测,检测条件如下:
[0014]
色谱柱填料为十八烷基硅烷键合硅胶;
[0015]
采用紫外检测器进行检测,检测波长为250nm~270nm;
[0016]
柱温为20℃~25℃;
[0017]
进样体积为5μl~15μl;
[0018]
流动相为磷酸盐缓冲液
‑
乙腈,所述磷酸盐缓冲液的浓度为0.01~0.02mol/l,所述磷酸盐缓冲液与所述乙腈的体积比为70:30~80:20。
[0019]
流速为0.6~1.2ml/min;
[0020]
洗脱方式为等度洗脱。
[0021]
优选的是,所述磷酸盐缓冲液为磷酸二氢钾缓冲液、磷酸二氢钠缓冲液中的至少一种;采用ph调节剂将所述磷酸盐缓冲液的ph值调节为5.5~6.5;所述色谱柱为waters xbridge shield rp18,所述色谱柱的长度为150mm。
[0022]
上述任一方案中优选的是,所述ph调节剂为氢氧化钠、氢氧化钾中的至少一种。
[0023]
上述任一方案中优选的是,所述制备富马酸丙酚替诺福韦供试品溶液包括如下步骤:
[0024]
利用稀释剂溶解富马酸丙酚替诺福韦制剂,过滤,取续滤液作为供试品溶液。
[0025]
上述任一方案中优选的是,所述制备富马酸丙酚替诺福韦对照品溶液包括如下步骤:
[0026]
利用稀释剂溶解富马酸丙酚替诺福韦对照品,制成每1ml含富马酸丙酚替诺福韦0.5~1.5mg的对照品溶液。
[0027]
上述任一方案中优选的是,所述稀释剂由稀释剂a和稀释剂b组成,所述稀释剂a为磷酸盐缓冲液,所述磷酸盐缓冲液的浓度为0.01~0.02mol/l,将所述磷酸盐缓冲液的ph值调节为5.5~6.5,所述稀释剂b为四氢呋喃的乙腈溶液,所述稀释剂b中四氢呋喃的体积分数为60%~80%。
[0028]
上述任一方案中优选的是,所述稀释剂a与所述稀释剂b的体积比为(90:10)~(99:1)。
[0029]
上述任一方案中优选的是,丙酚替诺福韦色谱峰的理论塔板数≥5000。
[0030]
上述任一方案中优选的是,在所述将所述供试品溶液与所述对照品溶液进行高效
液相色谱检测步骤中,检测条件如下:
[0031]
色谱柱为waters xbridge shield rp18;
[0032]
采用紫外检测器进行检测,检测波长为260nm;
[0033]
柱温为25℃;
[0034]
进样体积为10μl;
[0035]
流动相为磷酸二氢钾缓冲液
‑
乙腈,所述磷酸二氢钾缓冲液的浓度为0.02mol/l,采用ph调节剂将所述磷酸二氢钾缓冲液的ph值调节为6,所述磷酸二氢钾缓冲液与所述乙腈的体积比为70:30;
[0036]
流速为1ml/min;
[0037]
洗脱方式为等度洗脱。
[0038]
上述任一方案中优选的是,所述富马酸丙酚替诺福韦制剂为富马酸丙酚替诺福韦片、富马酸丙酚替诺福韦颗粒或富马酸丙酚替诺福韦胶囊。
[0039]
本发明提供的富马酸丙酚替诺福韦制剂中丙酚替诺福韦的检测方法能够有效分离丙酚替诺福韦及其相关物质,操作简便、线性关系良好、重复性良好、准确度好,能够提升对富马酸丙酚替诺福韦制剂的质量控制水平。
附图说明
[0040]
图1为按照实施例1提供的检测方法对空白辅料未降解溶液检测得到的高效液相色谱图;
[0041]
图2为实施例3中丙酚替诺福韦的浓度
‑
峰面积标准曲线图;
[0042]
图3为按照实施例4提供的检测方法对于0小时量取的供试品溶液检测得到的高效液相色谱图;
[0043]
图4为按照实施例4提供的检测方法对于0小时量取的供试品溶液检测得到的高效液相色谱图的峰表;
[0044]
图5为按照实施例4提供的检测方法对于0小时量取的对照品溶液检测得到的高效液相色谱图;
[0045]
图6为按照实施例4提供的检测方法对于0小时量取的对照品溶液检测得到的高效液相色谱图的峰表。
具体实施方式
[0046]
为了使本发明要解决的技术问题、技术方案及有益效果更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
[0047]
除另有定义外,以下实施例中所用的技术术语具有与本发明所属领域技术人员普遍理解的相同含义。以下实施例中所用的实验试剂,如无特殊说明,均为常规生化试剂,均可以通过市场上购买或者按照本领域常规方法合成制备;所述实验试剂用量,如无特殊说明,均为常规实验操作中试剂用量;所述实验方法,如无特殊说明,均为常规方法。
[0048]
本发明实施例提供了一种富马酸丙酚替诺福韦制剂中丙酚替诺福韦的检测方法,所述丙酚替诺福韦具有式ⅰ结构:
[0049][0050]
所述检测方法采用反相高效液相色谱法,包括如下步骤:
[0051]
(1)制备富马酸丙酚替诺福韦供试品溶液;
[0052]
(2)制备富马酸丙酚替诺福韦对照品溶液;
[0053]
(3)将所述供试品溶液与所述对照品溶液进行高效液相色谱检测,检测条件如下:
[0054]
色谱柱填料为十八烷基硅烷键合硅胶;
[0055]
采用紫外检测器进行检测,检测波长为250nm~270nm;
[0056]
柱温为20℃~25℃;
[0057]
进样体积为5μl~15μl;
[0058]
流动相为磷酸盐缓冲液
‑
乙腈,所述磷酸盐缓冲液的浓度为0.01~0.02mol/l(例如磷酸盐缓冲液的浓度可以为0.01mol/l、0.015mol/l或0.02mol/l等),所述磷酸盐缓冲液与所述乙腈的体积比为70:30~80:20(例如所述磷酸盐缓冲液与所述乙腈的体积比可以为70:30、75:25或80:20等)。如果将乙腈替换为甲醇,在专属性验证时,高温降解条件下,富马酸与相邻杂质的分离度小于1.5。
[0059]
流速为0.6~1.2ml/min(例如流速可以为0.6ml/min、0.8ml/min、1ml/min、或1.2ml/min等,优选地,流速为1ml/min);
[0060]
洗脱方式为等度洗脱。
[0061]
所述检测波长为250nm~270nm,例如检测波长可以为250nm、255nm、260nm、265nm或270nm等,丙酚替诺福韦在260nm附近处有最大吸收,因此,紫外检测器的检测波长设置为250nm~270nm之间,均可进一步提高检测的准确度。
[0062]
所述柱温为20℃~25℃,例如柱温可以为20℃、21℃、22℃、23℃或25℃等,柱温升高可加快分离过程,但因样品保留时间不稳将增加检测工作的麻烦,当柱温降低时,流动相黏度增加,分离过程时间会加长,会延长检测时间,因此控制柱温在20℃~25℃既可以确保检测结果的稳定性,也可以实现较快地检测富马酸丙酚替诺福韦制剂中丙酚替诺福韦,优选地,柱温为25℃。
[0063]
所述进样体积为5μl~15μl,例如进样体积可以为5μl、8μl、10μl、12μl、或15μl等,为了满足灵敏度要求且峰形最优,优选地,进样体积为10μl。
[0064]
洗脱方式为等度洗脱,操作方便,克服了梯度洗脱的缺点,例如采用梯度洗脱时对仪器要求较高,易产生气泡,影响基线噪声及漂移,同时会造成压力不稳定。
[0065]
根据本发明的实施例,将所述富马酸丙酚替诺福韦供试品溶液进行高效液相色谱检测分析,基于高效液相色谱检测结果,按照外标法确定所述富马酸丙酚替诺福韦供试品溶液中丙酚替诺福韦的含量,本发明提供的富马酸丙酚替诺福韦制剂中丙酚替诺福韦的检
测方法能够有效分离丙酚替诺福韦及其相关物质,操作简便、专属性强、分离度好、线性关系良好、重复性良好、准确度好,能够提升对富马酸丙酚替诺福韦制剂的质量控制水平。
[0066]
进一步地,所述磷酸盐缓冲液为磷酸二氢钾缓冲液、磷酸二氢钠缓冲液中的至少一种,采用ph调节剂将所述磷酸盐缓冲液的ph值调节为5.5~6.5,例如可以将所述磷酸盐缓冲液的ph值调节为5.5、6或6.5等,优选为6。
[0067]
进一步地,所述色谱柱为waters xbridge shield rp18,所述色谱柱的长度为150mm,色谱柱对富马酸丙酚替诺福韦制剂的检测具有重要影响,如果色谱柱(例如diamonsil c18(2)色谱柱(4.6mm
×
250mm,5μm))不合适,富马酸丙酚替诺福韦制剂中主成分及其相关物质的分离效果不理想,得到的峰形也并不理想,为了进一步地提高富马酸丙酚替诺福韦制剂中主成分及其相关物质的分离效果以及使得到的色谱图的峰形较好,优选地,所述色谱柱为waters xbridge shield rp18。
[0068]
进一步地,所述ph调节剂为氢氧化钠、氢氧化钾中的至少一种。
[0069]
进一步地,所述制备富马酸丙酚替诺福韦供试品溶液包括如下步骤:
[0070]
利用稀释剂溶解富马酸丙酚替诺福韦制剂,过滤,取续滤液作为供试品溶液。
[0071]
进一步地,所述制备富马酸丙酚替诺福韦供试品溶液包括如下步骤:
[0072]
取富马酸丙酚替诺福韦片10片,置500ml容量瓶中,用稀释剂溶解并稀释至刻度,过滤,取续滤液作为供试品溶液,所述富马酸丙酚替诺福韦片规格以丙酚替诺福韦计为25mg,即每片富马酸丙酚替诺福韦片含有丙酚替诺福韦25mg。
[0073]
进一步地,所述制备富马酸丙酚替诺福韦对照品溶液包括如下步骤:
[0074]
利用稀释剂溶解富马酸丙酚替诺福韦对照品,制成每1ml含富马酸丙酚替诺福韦0.5~1.5mg的对照品溶液,例如每1ml对照品溶液含富马酸丙酚替诺福韦可以为0.5mg、1mg或1.5mg等。
[0075]
进一步地,所述稀释剂由稀释剂a和稀释剂b组成,所述稀释剂a为磷酸盐缓冲液,所述磷酸盐缓冲液的浓度为0.01~0.02mol/l,将所述磷酸盐缓冲液的ph值调节为5.5~6.5,例如将所述磷酸盐缓冲液的ph值调节为5.5、6或6.5等,所述稀释剂b为四氢呋喃的乙腈溶液,所述稀释剂b中四氢呋喃的体积分数为60%~80%,例如所述稀释剂b中四氢呋喃的体积分数为60%、65%、70%、75%或80%等。
[0076]
进一步地,所述稀释剂a与所述稀释剂b的体积比为(90:10)~(99:1),例如稀释剂a与稀释剂b的体积比可以为90:10、92:8、94:6、96:4或99:1等。
[0077]
进一步地,丙酚替诺福韦色谱峰的理论塔板数≥5000。
[0078]
进一步地,在所述将所述供试品溶液与所述对照品溶液进行高效液相色谱检测步骤中,检测条件如下:
[0079]
色谱柱为waters xbridge shield rp18;采用紫外检测器进行检测,检测波长为260nm;柱温为25℃;进样体积为10μl;流动相为磷酸二氢钾缓冲液
‑
乙腈,所述磷酸二氢钾缓冲液的浓度为0.02mol/l,采用ph调节剂将所述磷酸二氢钾缓冲液的ph值调节为6,所述磷酸二氢钾缓冲液与所述乙腈的体积比为70:30;流速为1ml/min;洗脱方式为等度洗脱。
[0080]
本发明实施例提供了最优的检测条件,根据本发明实施例提供的检测条件对富马酸丙酚替诺福韦制剂中丙酚替诺福韦进行检测,该检测方法重复性好,专属性强,准确度好,分离效果好,线性良好,得到的色谱峰峰形优、基线平稳,能够提升对富马酸丙酚替诺福
韦制剂的质量控制水平。
[0081]
进一步地,所述富马酸丙酚替诺福韦制剂为富马酸丙酚替诺福韦片、富马酸丙酚替诺福韦颗粒或富马酸丙酚替诺福韦胶囊。
[0082]
进一步地,所述检测方法还包括获得标准曲线,所述标准曲线的获得是通过如下方式进行的:(1)利用前面所述的检测方法分别测定不同浓度的富马酸丙酚替诺福韦对照品,其中,丙酚替诺福韦的浓度为0.223mg/ml、0.357mg/ml、0.446mg/ml、0.535mg/ml、0.669mg/ml,(2)以峰面积为纵坐标,以丙酚替诺福韦的浓度为横坐标绘制标准曲线。由此,利用所述标准曲线,通过检测富马酸丙酚替诺福韦制剂中丙酚替诺福韦所对应的峰面积,按外标法以峰面积计算待测富马酸丙酚替诺福韦制剂中丙酚替诺福韦的含量。本发明实施例提供的检测方法测定含量时线性范围为0.223mg/ml~0.669mg/ml,线性范围浓度较高,配制时较为简便。
[0083]
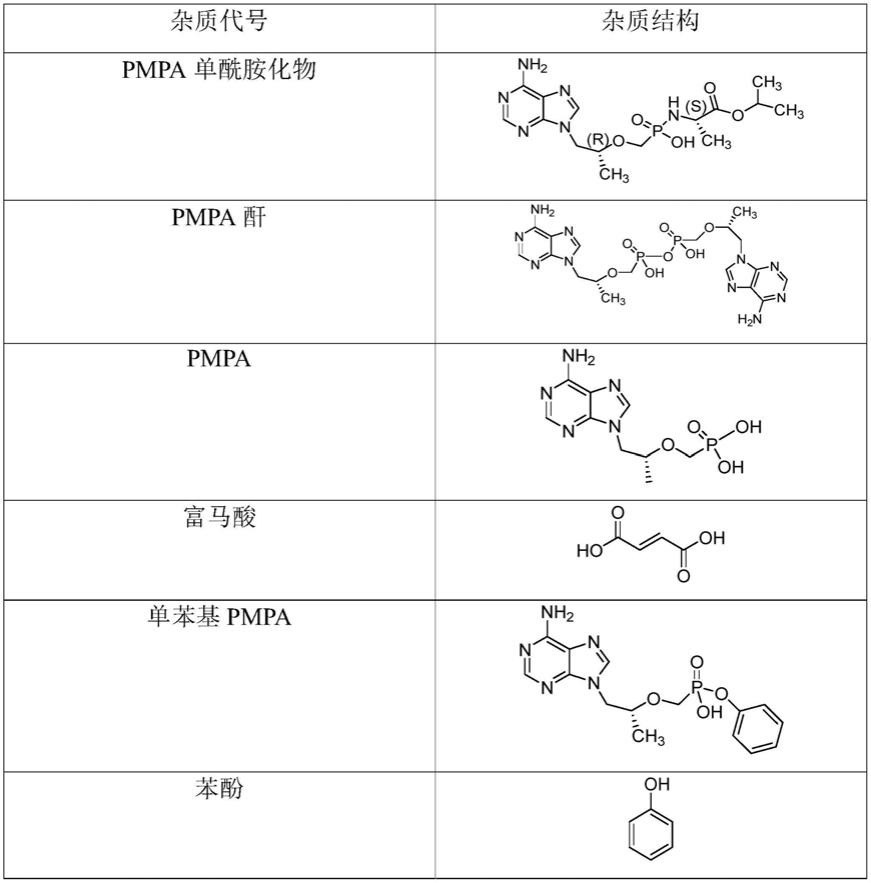
以下实施例中所涉及的杂质的化学结构式为:
[0084]
[0085][0086]
以下实施例中,富马酸丙酚替诺福韦片处方组成为:
[0087][0088][0089]
以下实施例中,采用的仪器如下:
[0090]
高效液相色谱仪:岛津hplc
‑
2030(紫外检测器);分析天平:sartorius cpa225d。
[0091]
实施例1专属性验证
[0092]
取富马酸丙酚替诺福韦片,采用高温、强酸、强碱、氧化、光照来加速本品的降解,以考察本色谱条件对主成分含量测定的专属性。
[0093]
色谱条件:色谱柱为waters xbridge shield rp18(150mm
×
4.6mm,3.5μm);流动相为0.02mol/l磷酸二氢钾溶液(用1mol/l氢氧化钾溶液调ph值至6)
‑
乙腈(体积比为70:30);流速为1.0ml/min;进样量为10μl;柱温为25℃;检测波长为260nm;洗脱方式为等度洗脱;运行时间为20min;丙酚替诺福韦色谱峰的理论塔板数≥5000。
[0094]
实验步骤:
[0095]
(1)稀释剂的配制:取磷酸二氢钾10.8833g,加水3000ml使溶解,混匀,用1mol/l氢氧化钾溶液调ph值至5.99,之后加水至4000ml,得到0.02mol/l磷酸盐缓冲液(ph6.0);分别量取乙腈300ml,四氢呋喃700ml,混匀,得到70%四氢呋喃的乙腈溶液;分别量取0.02mol/l磷酸盐缓冲液(ph6.0)1980ml,70%四氢呋喃的乙腈溶液20ml,混匀,得到稀释剂。
[0096]
(2)样品未降解溶液的配制:取富马酸丙酚替诺福韦片细粉约200mg(含丙酚替诺福韦25mg),精密称定,置50ml量瓶中,加稀释剂适量,40℃水浴恒温振荡器振摇60分钟使溶解,放冷至室温,用稀释剂稀释至刻度,摇匀,滤过,弃去初滤液5ml,取续滤液即得样品未降解溶液。
[0097]
空白辅料未降解溶液的配制:取空白辅料细粉约172mg,精密称定,置50ml量瓶中,加稀释剂适量,40℃水浴恒温振荡器振摇60分钟使溶解,放冷至室温,用稀释剂稀释至刻度,摇匀,滤过,弃去初滤液5ml,取续滤液即得空白辅料未降解溶液。
[0098]
样品酸降解溶液的配制:取富马酸丙酚替诺福韦片细粉约200mg(含丙酚替诺福韦25mg),精密称定,置50ml量瓶中,加0.1mol/l盐酸溶液1ml,室温放置10分钟,之后加0.1mol/l氢氧化钠溶液1ml中和,用稀释剂稀释至刻度,摇匀,滤过,弃去初滤液5ml,取续滤液即得样品酸降解溶液。
[0099]
空白辅料酸降解溶液的配制:取空白辅料细粉约172mg,精密称定,置50ml量瓶中,加0.1mol/l盐酸溶液1ml,室温放置10分钟,加0.1mol/l氢氧化钠溶液1ml中和,用稀释剂稀释至刻度,摇匀,滤过,弃去初滤液5ml,取续滤液即得空白辅料酸降解溶液。
[0100]
样品碱降解溶液的配制:取富马酸丙酚替诺福韦片细粉约200mg(含丙酚替诺福韦25mg),精密称定,置50ml量瓶中,加水5ml,加0.1mol/l氢氧化钠溶液1ml,室温放置1分钟,之后加0.1mol/l盐酸溶液1ml中和,用稀释剂稀释至刻度,摇匀,滤过,弃去初滤液5ml,取续滤液即得样品碱降解溶液。
[0101]
空白辅料碱降解溶液的配制:取空白辅料细粉约172mg,精密称定,置50ml量瓶中,加0.1mol/l氢氧化钠溶液1ml,室温放置1分钟,之后加0.1mol/l盐酸溶液1ml中和,用稀释剂稀释至刻度,摇匀,滤过,弃去初滤液5ml,取续滤液即得空白辅料碱降解溶液。
[0102]
样品氧化降解溶液的配制:取富马酸丙酚替诺福韦片细粉约200mg(含丙酚替诺福韦25mg),精密称定,置50ml量瓶中,加30%过氧化氢溶液1ml,室温放置6小时,加稀释剂稀释至刻度,摇匀,滤过,弃去初滤液5ml,取续滤液即得样品氧化降解溶液。
[0103]
空白辅料氧化降解溶液的配制:取空白辅料细粉约172mg,精密称定,置50ml量瓶中,加30%过氧化氢溶液1ml,室温放置6小时,加稀释剂稀释至刻度,摇匀,滤过,弃去初滤
液5ml,取续滤液即得空白辅料氧化降解溶液。
[0104]
样品高温降解溶液的配制:取富马酸丙酚替诺福韦片于105℃条件下放置7小时,取出,放冷至室温,取上述样品细粉约200mg(含丙酚替诺福韦25mg),精密称定,置50ml量瓶中,用稀释剂稀释至刻度,摇匀,滤过,弃去初滤液5ml,取续滤液即得样品高温降解溶液。
[0105]
空白辅料高温降解溶液的配制:取空白辅料于105℃条件下放置7小时,取出,放冷至室温,取所述空白辅料细粉约172mg,精密称定,置50ml量瓶中,用稀释剂稀释至刻度,摇匀,滤过,弃去初滤液5ml,取续滤液即得空白辅料高温降解溶液。
[0106]
样品光照降解溶液的配制:取富马酸丙酚替诺福韦片于4500
±
500lx光照条件下放置10天,取上述样品细粉约200mg(含丙酚替诺福韦25mg),精密称定,置50ml量瓶中,加稀释剂适量,40℃水浴恒温振荡器振摇60分钟使溶解,放冷至室温,用稀释剂稀释至刻度,摇匀,滤过,弃去初滤液5ml,取续滤液即得样品光照降解溶液。
[0107]
空白辅料光照降解溶液的配制:取富马酸丙酚替诺福韦片空白辅料于4500
±
500lx光照条件下放置10天,取上述空白辅料细粉约172mg,精密称定,置50ml量瓶中,加稀释剂适量,40℃水浴恒温振荡器振摇60分钟使溶解,放冷至室温,用稀释剂稀释至刻度,摇匀,滤过,弃去初滤液5ml,取续滤液即得空白辅料光照降解溶液。
[0108]
杂质定位溶液的配制:分别取pmpa单酰胺化物、pmpa酐、pmpa、富马酸、单苯基pmpa、苯酚、pmpa双酰胺化物、非对映异构体2、gs7339、非对映异构体1、联苯pmpa各适量,精密称定,分别用稀释剂溶解并稀释制成每1ml约含200μg/ml的溶液。
[0109]
(3)精密量取步骤(2)中制备的溶液各10μl,分别注入高效液相色谱仪,按照本实施例色谱条件进行检测,记录色谱图进行高效液相色谱分析,空白辅料未降解溶液的色谱图如图1所示,空白辅料酸降解溶液、空白辅料碱降解溶液、空白辅料氧化降解溶液、空白辅料高温降解溶液和空白辅料光照降解溶液的色谱图与空白辅料未降解溶液的色谱图类似。测得的各杂质的保留时间如表1所示,富马酸丙酚替诺福韦片破坏试验的专属性考察结果如表2所示。
[0110]
表1
[0111]
名称保留时间(分钟)pmpa单酰胺化物2.160pmpa酐2.161富马酸2.198pmpa2.256单苯基pmpa2.576苯酚9.856pmpa双酰胺化物10.709非对映异构体211.894主峰(丙酚替诺福韦色谱峰)12.209gs733913.271非对映异构体114.076联苯pmpa18.701
[0112]
表2
[0113]
[0114][0115]
在本实施例色谱条件下,所有空白辅料溶液均未出现色谱峰,因此空白辅料和稀释剂不干扰丙酚替诺福韦检出;各杂质定位溶液保留时间与主成分均不一致,各杂质不干扰丙酚替诺福韦检出。本品在酸降解试验条件下产生2个杂质,碱降解试验条件下产生1个杂质,高温降解试验条件下产生4个杂质,氧化降解试验条件下产生2个杂质,但各杂质与主成分(丙酚替诺福韦)之间的分离度均大于1.5,丙酚替诺福韦峰理论塔板数均大于5000,且峰纯度符合要求,表明本实施例提供的富马酸丙酚替诺福韦片含量测定方法专属性良好。
[0116]
实施例2定量限
[0117]
色谱条件:同实施例1色谱条件。
[0118]
实验步骤:
[0119]
(1)稀释剂的配制:配制方法同实施例1中稀释剂的配制。
[0120]
(2)定量限贮备液的配制:取富马酸丙酚替诺福韦对照品约50mg,精密称定,置50ml量瓶中,用稀释剂溶解并稀释至刻度,摇匀,精密量取5ml,置10ml量瓶中,用稀释剂溶解并稀释至刻度,摇匀,得到混合液,精密量取1ml该混合液,置100ml量瓶中,用稀释剂溶解并稀释至刻度,摇匀,得到定量限贮备液。
[0121]
定量限溶液的配制:精密量取定量限贮备液1ml,置100ml量瓶中,用稀释剂溶解并稀释至刻度,摇匀。(平行配制6份)
[0122]
(3)取步骤(2)制备的6份定量限溶液各10μl,分别注入液相色谱仪,记录色谱图并进行高效液相色谱分析,当信噪比(s/n)约为(10:1)时,丙酚替诺福韦的浓度为定量限。定量限考察试验结果如表3所示,rsd为相对标准偏差。
[0123]
表3
[0124][0125][0126]
由表3可知,丙酚替诺福韦的定量限为44.57ng/ml,相当于供试品浓度(0.5mg/ml)的0.009%,符合药典中对定量限的规定。
[0127]
实施例3线性与范围
[0128]
色谱条件:同实施例1色谱条件。
[0129]
实验步骤:
[0130]
(1)稀释剂的配制:与实施例1中稀释剂的配制相同。
[0131]
(2)线性贮备液的配制:取富马酸丙酚替诺福韦对照品约50mg,精密称定,置50ml量瓶中,用稀释剂溶解并稀释至刻度,摇匀,得到线性贮备液。
[0132]
线性溶液的配制:分别精密量取线性贮备液7.5ml、6.0ml、5.0ml、4.0ml、2.5ml,分
别置不同10ml量瓶中,加稀释剂稀释至刻度,摇匀,分别作为线性150%、120%、100%、80%、50%浓度的溶液。
[0133]
(3)精密量取步骤(2)制备的不同浓度的线性溶液各10μl,分别注入液相色谱仪,记录色谱图并进行高效液相色谱分析。以丙酚替诺福韦浓度(c)为横坐标(x轴),以丙酚替诺福韦对应的色谱峰的峰面积(a)为纵坐标(y轴),进行线性回归分析,结果如表4和图2所示。
[0134]
表4
[0135][0136]
由表4可知,丙酚替诺福韦在0.223mg/ml~0.669mg/ml(相当于供试品浓度的50%~150%)浓度范围内,线性方程为y=17070799.5000x+297437.2310,相关系数r为1.0000,大于0.9990,表明丙酚替诺福韦浓度在0.223mg/ml~0.669mg/ml范围内进样,峰面积与浓度呈良好的线性关系。
[0137]
实施例4溶液稳定性
[0138]
色谱条件:同实施例1色谱条件。
[0139]
实验步骤:
[0140]
(1)稀释剂的配制:与实施例1中稀释剂的配制相同。
[0141]
(2)对照品溶液的配制:取富马酸丙酚替诺福韦对照品约28mg,精密称定,置50ml量瓶中,用稀释剂溶解并稀释至刻度,摇匀,得到对照品溶液。
[0142]
供试品溶液的配制:取富马酸丙酚替诺福韦片10片,置500ml量瓶中,加适量稀释剂振摇60分钟,取出,冷却至室温,用稀释剂稀释至刻度,摇匀,滤过,弃去初滤液5ml,取续滤液,得到供试品溶液。
[0143]
(3)将步骤(2)制备的供试品溶液和对照品溶液分别于室温下放置24小时,分别于0、1、3、5、7、9、11、13、15、17、24小时精密量取供试品溶液与对照品溶液各10μl,分别注入液相色谱仪,记录色谱图并进行高效液相色谱分析,于0小时量取供试品溶液进行色谱分析的色谱图和对应的峰表如图3和图4所示,于0小时量取对照品溶液进行色谱分析的色谱图和对应的峰表如图5和图6所示,溶液稳定性考察试验结果如表5所示。
[0144]
表5
[0145][0146][0147]
由表5可知对照品溶液、供试品溶液于室温下放置24小时,峰面积rsd值分别为0.16%、0.22%,均小于1.0%,表明对照品溶液、供试品溶液在室温下放置24小时稳定。
[0148]
实施例5精密度验证
[0149]
色谱条件:同实施例1色谱条件。
[0150]
实验步骤:
[0151]
(1)对照品溶液的配制:与实施例4中对照品溶液的配制相同。
[0152]
(2)精密量取对照品溶液10μl,注入液相色谱仪,连续进样6次,记录色谱图并进行高效液相色谱分析,精密度考察试验结果如表6所示。
[0153]
表6
[0154]
[0155][0156]
对照品溶液连续进样6次,保留时间与峰面积的rsd值分别为0.05%、0.04%,均小于1.0%,表明仪器进样精密度良好。
[0157]
实施例6重复性验证
[0158]
色谱条件:同实施例1色谱条件。
[0159]
实验步骤:
[0160]
(1)稀释剂的配制:与实施例1中稀释剂的配制相同。
[0161]
(2)对照品溶液的配制:与实施例4中对照品溶液的配制相同,平行配制2份。
[0162]
供试品溶液的配制:与实施例4中供试品溶液的配制相同,平行配制6份。
[0163]
(3)精密量取对照品溶液与供试品溶液各10μl,分别注入液相色谱仪,记录色谱图并进行高效液相色谱分析,按外标法以峰面积计算丙酚替诺福韦含量,重复性考察试验结果如表7所示,含量(%)计算公式为:
[0164][0165]
式中:测得量为按外标法以峰面积计算每片富马酸丙酚替诺福韦片中丙酚替诺福韦的含量;标示量为每片富马酸丙酚替诺福韦片中规定的丙酚替诺福韦的含量,即25mg。
[0166]
表7
[0167][0168][0169]
由表7可知,平行配制的6份供试品溶液,丙酚替诺福韦含量均在95%~105%范围内,平均值为102.6%,rsd值为0.49%,小于1.0%,表明富马酸丙酚替诺福韦片含量测定方法重复性良好。
[0170]
实施例7准确度验证
[0171]
色谱条件:同实施例1色谱条件。
[0172]
实验步骤:
[0173]
(1)稀释剂的配制:与实施例1中稀释剂的配制相同。
[0174]
(2)对照品溶液的配制:与实施例4中对照品溶液的配制相同。
[0175]
80%供试品溶液的配制:取空白辅料约172mg与富马酸丙酚替诺福韦对照品约22.4mg,精密称定,置同一50ml量瓶中,用稀释剂溶解并稀释至刻度,摇匀,滤过,弃去初滤液5ml,取续滤液,得到80%供试品溶液,平行配制3份。
[0176]
100%供试品溶液的配制:取空白辅料约172mg与富马酸丙酚替诺福韦对照品约28mg,精密称定,置同一50ml量瓶中,用稀释剂溶解并稀释至刻度,摇匀,滤过,弃去初滤液5ml,取续滤液,得到100%供试品溶液,平行配制3份。
[0177]
120%供试品溶液的配制:取空白辅料约172mg与富马酸丙酚替诺福韦对照品约33.6mg,精密称定,置同一50ml量瓶中,用稀释剂溶解并稀释至刻度,摇匀,滤过,弃去初滤液5ml,取续滤液,得到120%供试品溶液,平行配制3份。
[0178]
(3)精密量取步骤(2)中各溶液各10μl,分别注入液相色谱仪,记录色谱图并进行高效液相色谱分析,按外标法以峰面积计算丙酚替诺福韦含量,按下式计算回收率(%):
[0179][0180]
准确度考察试验结果如表8所示。
[0181]
表8
[0182][0183]
由表8可知,在相当于供试品浓度的80%~120%浓度范围内,9份样品回收率均在98%~102%之间,平均值为100.2%,rsd值为0.27%,小于2%,表明本发明实施例提供的富马酸丙酚替诺福韦片含量测定方法准确度良好。
[0184]
实施例8富马酸丙酚替诺福韦片中丙酚替诺福韦的检测
[0185]
色谱条件:同实施例1色谱条件。
[0186]
实验步骤:
[0187]
(1)稀释剂的配制:与实施例1中稀释剂的配制相同。
[0188]
(2)对照品溶液的配制:与实施例4中对照品溶液的配制相同,平行配制2份。
[0189]
供试品溶液的配制:与实施例4中供试品溶液的配制相同。
[0190]
空白辅料溶液的配制:与实施例1中空白辅料未降解溶液的配制相同。
[0191]
(3)精密量取对照品溶液、供试品溶液和空白辅料溶液各10μl,分别注入液相色谱仪,记录色谱图并进行高效液相色谱分析,丙酚替诺福韦色谱峰的理论塔板数>5000,供试品溶液色谱图中丙酚替诺福韦,应与对照品溶液色谱图中丙酚替诺福韦保留时间一致,空白辅料溶液色谱图无与对照品溶液色谱图中丙酚替诺福韦色谱峰相同的峰出现,即空白辅料溶液无干扰。丙酚替诺福韦浓度在0.223mg/ml~0.669mg/ml范围内进样,峰面积与浓度呈良好的线性关系,按外标法以峰面积计算丙酚替诺福韦含量,测得平均每片富马酸丙酚替诺福韦片中丙酚替诺福韦的重量为25.5mg。
[0192]
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1