一种对尿液中的合成卡西酮类毒品进行快速定量的方法与流程
1.本发明属于法医毒物分析鉴定领域,具体涉及一种利用磁分散固相萃取法(mdspe)结合实时直接分析高分辨质谱法(dart
‑
hrms)对尿液中的合成卡西酮类毒品进行快速定量的方法。
背景技术:
2.新精神活性物质(nps)是在管制毒品的结构基础上设计和非法制造出来的毒品衍生物,用于模仿管控毒品的精神作用,逃脱刑法制裁。截止2019年,所报道的新精神活性物质已经超过800种,涉及全球110多个国家且依然在不断增加,给全球公共安全带来极大的挑战,被描述为“全球范围内的流行病”。
3.根据联合国毒品和犯罪问题办公室的最新报告,合成卡西酮类与合成大麻素类是全世界缉获的nps最多的类别。有数据表明,卡西酮类与大麻相比能更快的出现成瘾性和戒断综合征。在中国,已有188种新精神活性物质被列入管制,其中合成卡西酮类毒品共有57种。
4.合成卡西酮类毒品拥有共同的骨架结构,n取代形成仲胺、叔胺和吡咯烷基;
ɑ
‑
c被烷基取代及苯环被烷基、亚甲二氧基和卤素取代形成各种合成卡西酮类衍生物。
5.(4
‑
氯苯基)
‑2‑
(n
‑
吡咯烷基)
‑1‑
戊酮(简写为4
‑
cl
‑
α
‑
pvp),2
‑1‑
(4
‑
甲基苯基)
‑2‑
甲氨基
‑1‑
戊酮(简写为4
‑
mpd)和3
‑1‑
[2
‑
(5,6,7,8
‑
四氢萘基)]
‑2‑
(n
‑
吡咯烷基)
‑1‑
戊酮(简写为β
‑
th
‑
naphyrone)涵盖了常见的卡西酮类衍生物的取代情况,但目前没有报道关于β
‑
th
‑
naphyrone和4
‑
mpd的定量方法,4
‑
cl
‑
α
‑
pvp定量也仅有少量报道。
[0006]
毒品领域常见的人体检材有血液、尿液、唾液、毛发等,尿液因其易收集无伤害而常被人们所使用,且合成卡西酮类毒品在尿液中原体存在较多。现有尿液中合成卡西酮类物质的定量方法大多为气相色谱
‑
质谱联用(gc
‑
ms)和液相色谱
‑
质谱联用(lc
‑
ms),但是经过色谱方法都会存在耗时长的缺陷,且多数gc
‑
ms还需要衍生化。antunes m等(determination of selected cathinones in blood by solid
‑
phase extraction and gc
‑
ms.j anal toxicol.2021mar 12;45(3):233
‑
242)使用固相萃取法(spe)萃取血中4
‑
cl
‑
α
‑
pvp时间大于55min;使用gc
‑
ms定量4
‑
cl
‑
α
‑
pvp在10.173min出峰,检出限和定量限分别为5ng/ml和25ng/ml。wang p等(qualitative and quantitative analysis of cathinones in human urine by spe
‑
gc
‑
ms.fa yi xue za zhi.2018jun;34(6):606
‑
610)研究了4种合成卡西酮类物质的检测,其前处理方式为spe且衍生化,前处理用时大于26min,检测方式为gc
‑
ms,检测用时10min,lod和lloq为2ng/ml和25ng/ml,前处理及检测耗时均过长,检测限较低。因此,迫切需要一种简单、快速、准确的方法去定量合成卡西酮类毒品。
[0007]
自从2005年cody rb等(versatile new ion source for the analysis of materials in open air under ambient conditions.anal chem.2005apr15;77(8):2297
‑
302)提出“用于环境条件下露天材料分析的多功能新型离子源”后,实时直接分析便
因其在常温常压下对样品进行直接分析而被人们喜爱,之前多被用于快速筛查。尽管实时直接分析可在1min内快速出结果,非常吸引人们在定量上对其进行探索,但是由于dart
‑
ms灵敏度比lc
‑
ms灵敏度低,与gc
‑
ms灵敏度相似,容易受到信号波动的影响,且由于尿液中存在高浓度的尿素和肌酐,严重干扰电离过程,导致灵敏度低和再现性差,因此难以通过dart
‑
ms测定尿液中的合成卡西酮,目前尚无dart
‑
ms定量合成卡西酮类毒品的报道。
[0008]
现有的尿液中合成卡西酮类毒品前处理方式有固相萃取法(spe)和液液萃取法(lpe)等,固相萃取法和液液萃取法通常需要蒸发的步骤去除溶剂再进一步富集,而这一过程繁琐,需要大量的溶剂且非常耗费时间,如takaya murakami等(molecularly imprinted polymer solid
‑
phase extraction of synthetic cathinones from urine and whole blood samples.j sep sci.2018dec;41(24):4506
‑
4514)采用盐析辅助spe萃取尿液中的11种合成卡西酮类物质需要经过三次溶剂洗涤,干燥后再用醋酸、乙腈溶解。enrico gerace等(determination of several synthetic cathinones and an amphetamine
‑
like compound in urine by gas chromatography with mass spectrometry.method validation and application to real cases.j sep sci.2019apr;42(8):1577
‑
1584)采用碱性条件下液液萃取,取有机层干燥后衍生化,提取人尿液中18种合成卡西酮类物质,步骤繁琐,耗时以小时起步。
[0009]
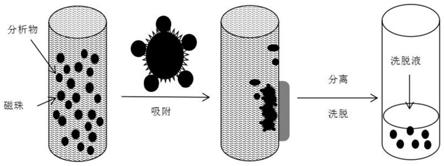
传统的固相萃取法还需要平衡活化柱子,基于磁性颗粒的磁分散固相萃取法与传统的固相萃取法原理相似,通过范德华力、氢键及相似相溶的特点吸附待测物,但与spe不同的是,磁性颗粒是分散在待测基质中的,这表明所有的颗粒会同时吸附待测物,参见图1,大量磁性颗粒分散在基质中快速捕捉待测物,然后通过施加外加磁场,使吸附在磁性颗粒上的待测物与基质快速分离,之后通过洗脱剂将待测物从磁性颗粒上洗脱下来。这一过程不需要柱分离技术和离心,耗材少,操作简单,不需要蒸发和衍生化,提取时间短,然而对于不同的处理对象,分析对象与磁珠表面的官能团相匹配时才可使用。
[0010]
磁分散固相萃取法已经广泛的用于金属领域和生物活性物质领域,但在毒品小分子领域的应用只有很少报道,lu q等人(graphene oxide
‑
fe3o4nanocomposite magnetic solid phase extraction followed by uhplc
‑
ms/ms for highly sensitive determination of eight psychoactive drugs in urine samples.talanta.2020jan 1)用氧化石墨烯
‑
fe3o4纳米复合磁性吸附剂萃取尿液中吗啡、苯丙胺;yang f等(magnetic dispersive solid
‑
phase extraction based on modified magnetic nanoparticles for the detection of cocaine and cocaine metabolites in human urine by high
‑
performance liquid chromatography
‑
mass spectrometry.j sep sci.2017nov;40(21):4234
‑
424)用改良的fe3o4合成纳米颗粒萃取尿液中可卡因及其代谢物,然而该两种方法中的磁珠类型难以应用至尿液中合成卡西酮类毒品的定量检测中。
技术实现要素:
[0011]
本发明的目的在于提供一种对尿液中的合成卡西酮类毒品进行快速定量的方法,通过磁分散固相萃取法(mdspe)提取尿液中的合成卡西酮类毒品,结合实时直接分析高分辨质谱法(dart
‑
hrms)进行快速定量,进样后可在1分钟之内得出定量结果,待测物在各条件下稳定性良好,相关系数大于0.99,精密度及准确度偏差均小于15%,检测快速、便捷,能
够为法医毒物分析中快速定量合成卡西酮类毒品提供新的、有效的技术支持。
[0012]
为了达到上述目的,本发明提供如下技术方案:
[0013]
一种对尿液中的合成卡西酮类毒品进行快速定量的方法,包括以下步骤:
[0014]
1)配制工作溶液
[0015]
称取合成卡西酮类毒品标准品,配制成标准品储备液,用空白尿液逐级稀释成浓度梯度分布的标准工作溶液;以甲卡西酮
‑
d3和skf525a作为内标物质,配制成内标标准溶液;
[0016]
2)磁分散固相萃取
[0017]
取标准工作溶液,分别加入内标标准溶液,进行磁分散固相萃取,得到含内标物质的标准工作溶液洗脱液;
[0018]
所述磁分散固相萃取中的磁珠为二乙烯基苯与乙烯基吡咯烷酮以2.8
‑
3.3:1的比例制成的疏水性磁珠;
[0019]
3)制作标准曲线
[0020]
将步骤2)中获得的标准工作溶液洗脱液进行dart
‑
hrms检测,获得梯度浓度下每个分析物和内标物质的峰面积数据,制作分析物和内标物质峰面积比值与浓度之间的标准曲线;
[0021]
4)样品定量
[0022]
取待测尿液样品,加入内标标准溶液,进行磁分散固相萃取,得到含内标物质的待测样品洗脱液,将该含内标物质的待测样品洗脱液进行dart
‑
hrms检测,获得待测样品与内标物质的峰面积比值,根据步骤3)确定的标准曲线获得定量结果。
[0023]
进一步,步骤1)中,所述标准溶液为混合标准溶液,其中含有两种以上的合成卡西酮类毒品。
[0024]
优选地,所述混合标准溶液中每种合成卡西酮类毒品的浓度从0.04
‑
20μg/ml之间梯度分布。
[0025]
优选地,所述标准工作溶液的浓度梯度范围为0.2
‑
100ng/ml,待测样品中加入内标物质的浓度范围为50
‑
66ng/ml。
[0026]
又,步骤2)及步骤4)中,所述磁分散固相萃取的过程是:向相应样品中加入ph为7的缓冲溶液,离心后取上清液,加入磁珠,混匀,后入磁珠吸附盘静止1min,去尿液,加纯水震荡清洗,再入磁珠吸附盘静止1min,去水加洗脱剂洗脱。
[0027]
进一步,步骤2)及步骤4)中,所述ph为7的缓冲溶液由nah2po4和naoh配制成,磁珠浓度为50mg/ml,体积为100
‑
300μl,混匀时间为2
‑
6min,纯水震荡清洗时间为1
‑
3min,洗脱时间为1
‑
3min。
[0028]
优选地,步骤2)及步骤4)中,所述洗脱剂为0.1%甲酸的乙腈溶液,体积为100
‑
300μl。
[0029]
又,步骤2)及步骤4)中,样品溶液、磁珠和缓冲液的体积比为1:0.1
‑
0.3:0.5。
[0030]
进一步,步骤3)和步骤4)中,相应的洗脱液直接进样检测,dart
‑
hrms检测条件为:dart执行正离子模式,氦气流速为3.5l/min,运行温度350
‑
400℃,进样量1.5
‑
2μl,进样速度0.4mm/s,栅极电压为150
‑
200v;ms以全扫描模式执行,毛细管温度为320℃,分辨率设置为70000;质量范围是m/z 50
‑
750。
[0031]
优选地,所述合成卡西酮类毒品为4
‑
cl
‑
α
‑
pvp、4
‑
mpd和/或β
‑
th
‑
naphyrone。
[0032]
本发明中对尿液中的合成卡西酮类毒品的快速定量方法用于人或大鼠尿液测定。
[0033]
本发明中,以甲卡西酮
‑
d3和skf525a作为内标,nah2po4/naoh(ph7)作为缓冲液,二乙烯基苯与乙烯基吡咯烷酮以2.8
‑
3.3:1的比例组成疏水性磁珠,用于磁分散固相萃取,洗脱后,洗脱液直接移至12dip
‑
it玻璃毛细管上由dart
‑
hrms检测,可在1分钟之内得出定量结果,各待测物在出峰位点均不存在干扰,高浓度后无残留,不影响后续实验。
[0034]
本发明中,针对合成卡西酮类毒品选取的两种内标物质甲卡西酮
‑
d3和skf525a,既满足在dart进样时与待测物同条件电离,还有较高的特性被所述磁珠提取,提高了数据的精密度和仪器的稳定性,克服了实时直接分析质谱检测的低重现性。本发明中,内标物质的用量明显影响数据稳定性,比如甲卡西酮
‑
d3浓度过低时,其与4
‑
cl
‑
α
‑
pvp和4
‑
mpd的峰面积比值不稳定,且甲卡西酮
‑
d3浓度越低,数据的波动越大;而甲卡西酮
‑
d3浓度为50ng/ml
‑
66ng/ml时,比值非常稳定,精密度rsd在15%以内。由于毒品的内标成本较高,因此,在满足实验条件的前提下尽可能减少内标的用量,本发明选择甲卡西酮
‑
d3、skf525a为50
‑
66ng/ml的浓度。
[0035]
本发明在dart
‑
hrms检测时,以待测物响应作为依据优化设备运行参数,最优运行温度为350
‑
400℃,进样量为1.5
‑
2μl,最优进样速度为0.4mm/s,最优栅极电压为150
‑
200v。
[0036]
在本发明的磁分散固相萃取中,磁珠负责将待测物从尿液中吸附出来,混匀仪360
°
慢速旋转,使尿液中的待测物充分的被吸附在磁珠上。去除尿液后经过水洗去除部分杂质,此时待测物依旧停留在磁珠上,通过结合能力较强的洗脱剂将待测物从磁珠上洗脱下来后进样。
[0037]
磁珠表面的官能团对于合成卡西酮类毒品的吸附是至关重要的,合成卡西酮类毒品的共同结构是都有一个苯环(非极性)和一个氮原子(极性),因此,本发明选择了不同比例的疏水官能团(二乙烯基苯)与亲水性官能团(乙烯基吡咯烷酮)形成亲水性、疏水性和中性的磁珠,可以同时与极性和非极性物质相互作用。实验表明,二乙烯基苯和乙烯基吡咯烷酮2.8
‑
3.3:1时(即疏水性磁珠),合成卡西酮类毒品的回收率最佳,对4
‑
cl
‑
α
‑
pvp、4
‑
mpd和β
‑
th
‑
naphyrone在不同浓度的回收率cv分别为6.99%、9.87%、4.47%,三者可以单独定量,也可同时定量;因此,本发明选择疏水性磁珠作为最终吸附剂,与合成卡西酮类毒品具有很好的匹配度,所得响应强,回收率高。
[0038]
本发明中,对于尿液样品来说,离心和缓冲液体系都是不可缺少的条件,因为个体之间存在差异,新鲜尿液的ph值通常在4.5至7.9之间,且会因存放时间条件等不同而发生变化,因此使用缓冲体系,可以尽可能的缩小不同来源尿液的差距,而离心则可以去除尿液中的沉淀颗粒。
[0039]
本发明的磁珠对于合成卡西酮类毒品的吸附在ph7缓冲液下效果最好,在酸性和碱性条件下存在不同程度的抑制,所有待测物在酸性缓冲液下响应是最低的,因此,本发明选择ph7的缓冲液。
[0040]
本发明的磁分散固相萃取中,洗脱剂需要比磁珠有更强的争夺合成卡西酮类待测物的能力,还要考虑待测物与内标峰面积的比值稳定性,对于甲醇、乙醇、乙腈和乙酸乙酯这四种洗脱剂,乙腈和乙酸乙酯的效果较好,但在乙酸乙酯中分析物与内标的峰面积比值不稳定。质谱检测为正离子模式,推测在酸性条件下会促进待测物离子化,因此加入0.1%
nanoparticles for the detection of cocaine and cocaine metabolites in human urine by high
‑
performance liquid chromatography
‑
mass spectrometry.j sep sci.2017nov;40(21):4234
‑
424)中的磁珠制备方法制得;1
‑
(4
‑
氯苯基)
‑2‑
(n
‑
吡咯烷基)
‑1‑
戊酮(4
‑
cl
‑
α
‑
pvp)、1
‑
(4
‑
甲基苯基)
‑2‑
甲氨基
‑1‑
戊酮(4
‑
mpd)、1
‑
[2
‑
(5,6,7,8
‑
四氢萘基)]
‑2‑
(n
‑
吡咯烷基)
‑1‑
戊酮(β
‑
th
‑
naphyrone)购买自上海原思标物科技有限公司;甲卡西酮
‑
d3盐酸盐购自cerilliant公司,普罗地芬盐酸盐(skf525a)购自sigma
‑
aldrich公司。
[0052]
实验中所用设备有:混匀仪be
‑
1100,kylin
‑
bell实验室;离心机,德国eppendorf公司;便携式ph计,wtw公司;exactive plus高分辨质谱仪,美国thermo fisher公司,实时直接分析离子源svp
‑
201,美国ion sense公司。
[0053]
实施例1利用磁分散固相萃取结合dart
‑
hrms对尿液中的4
‑
cl
‑
α
‑
pvp、4
‑
mpd、β
‑
th
‑
naphyrone进行快速定量的方法
[0054]
1)配制工作溶液
[0055]
分别精密称取4
‑
cl
‑
α
‑
pvp、4
‑
mpd、β
‑
th
‑
naphyrone、skf525a,置于棕色试剂瓶中用甲醇配制成浓度为1.00mg/ml储备液在4℃下储存备用,内标物质甲卡西酮
‑
d3(用乙腈)和skf525a(用甲醇)各稀释成10μg/ml储备液置于棕色试剂瓶中在4℃下储存备用。
[0056]
分别取1.00mg/ml上述储备液配制成含4
‑
mpd、β
‑
th
‑
naphyrone各0.04μg/ml、0.1μg/ml、1μg/ml、4μg/ml、10μg/ml、15μg/ml、20μg/ml及含4
‑
cl
‑
α
‑
pvp各0.1μg/ml、0.2μg/ml、1μg/ml、4μg/ml、10μg/ml、15μg/ml、20μg/ml的混合标准溶液,在4℃下储存备用,后用空白尿液逐级稀释成浓度梯度分布的标准工作溶液。
[0057]4‑
cl
‑
α
‑
pvp的质控样品浓度为:0.5ng/ml、1ng/ml、50ng/ml、75ng/ml;4
‑
mpd和β
‑
th
‑
naphyrone各自的质控样品浓度为:0.2ng/ml、0.5ng/ml、50ng/ml、75ng/ml。
[0058]
2)仪器参数优化
[0059]
将步骤1)中获得的混合标准溶液进行dart
‑
hrms检测:
[0060]
待测样品溶液直接由12dip
‑
it尖端扫描仪自动进样器采样,dart执行正离子模式,氦气流速为3.5l/min,毛细管温度为320℃;分辨率设置为70000;质量范围是m/z 50
‑
750。
[0061]
xcalibur软件(2.1版,thermo fisher scientific,美国加利福尼亚州圣何塞)控制运行。
[0062]
在不同条件下优化了运行温度、进样速度和栅极电压,在150℃
‑
450℃内以50℃为阶段选择运行温度,在0.2
‑
1mm/s内以0.2mm/s为阶段选择进样速度,在100
‑
300v,以50v为阶梯优化栅极电压,结果如图2所示,对于运行温度,β
‑
th
‑
naphyrone在400℃下达到最优,4
‑
cl
‑
α
‑
pvp和4
‑
mpd在350℃下达到最优,考虑到整体检测4
‑
cl
‑
α
‑
pvp响应最低,因此,本实施例以4
‑
cl
‑
α
‑
pvp为参考选择350℃为最优温度。
[0063]
对于进样速度,参见图3,三种待测物在0.4mm/s下响应均达到最好,0.6mm/s及之后的速度对4
‑
cl
‑
α
‑
pvp和4
‑
mpd干扰较大,因此选择0.4mm/s为最优进样速度。对于栅极电压,参见图4,150v和200v时待测物响应明显强于其他电压,150v和200v待测物响应相近。
[0064]
最优的dart
‑
hrms检测条件为:dart执行正离子模式,氦气流速为3.5l/min,运行温度350℃,进样量2μl,进样速度0.4mm/s,栅极电压为200v。
[0065]
3)磁分散固相萃取
[0066]
分别取上述梯度浓度的标准工作溶液,用空白尿液稀释,得到梯度加标样品各1ml,分别进行磁分散固相萃取,具体是:向每个样品中加入两种内标溶液各5μl及nah2po4/naoh(0.2m,ph7)缓冲液500μl,离心3min取上清液,加入磁珠(50mg/ml,100μl),混匀2min,入磁珠吸附盘静止1min,去尿液,加1ml纯水震荡清洗2min,入磁珠吸附盘静止1min,去水加100μl洗脱剂,洗脱1min,移出后得到梯度浓度的含内标物质的洗脱液;
[0067]
所述磁珠为二乙烯基苯与乙烯基吡咯烷酮以3:1的比例制成的疏水性磁珠;
[0068]
其中,考察了甲醇、乙醇、乙腈和乙酸乙酯四种洗脱剂,乙腈和乙酸乙酯的效果较好,但在乙酸乙酯中分析物与内标的峰面积比值不稳定。质谱检测为正离子模式,推测在酸性条件下会促进待测物离子化,因此又考察0.1%甲酸
‑
乙腈的洗脱效果,实验结果如图5所示,整体响应中0.1%甲酸
‑
乙腈效果最优,因此选择0.1%甲酸
‑
乙腈作为最优洗脱剂。
[0069]
对磁珠用量、混匀时间及洗脱时间采用均匀设计做1因素3水平和2因素6水平的拟水平设计,设计方案见表1。
[0070]
表1拟水平设计u6(3
×
62)
[0071][0072]
所得模型方差分析结果为p(4
‑
cl
‑
α
‑
pvp)=0.233,p(4
‑
mpd)=0.316,p(β
‑
th
‑
naphyrone)=0.564,均大于0.05,表明所得模型不成立,即在所测的条件下,各影响因素和各待测物响应不存在明显的线性关系,因此可认定各影响因素的值可在该条件范围内均可设定,本实施例选择磁珠用量100μl、混匀时间2min、洗脱时间1min。
[0073]
4)制作标准曲线
[0074]
将步骤3)中获得的标准品洗脱液以步骤2)的条件进行dart
‑
hrms检测,获得梯度浓度下每个待测物与内标物质的峰面积,以待测物和内标物质的峰面积比值与浓度关系制作标准曲线,并确定lod和lloq。
[0075]
5)方法验证
[0076]
根据欧洲药品管理局生物分析方法验证指南进行以下方法学验证:
[0077]
将质控样品以步骤1)中质控样品浓度,用步骤3)相同的方法进行磁分散固相萃取,获得的质控样品洗脱液进行方法学验证,对选择性和残留、准确度和精密度、稀释可靠性、基质效应和回收率、稳定性进行验证;
[0078]
5.1选择性和残留
[0079]
使用实验室志愿者提供的6个空白基质评价干扰,通过在进样100ng/ml的空白尿液加标样品(n=6)后进样空白样品来估计残留。
[0080]
5.2检测限(lod)、定量下限(lloq)
[0081]
分别进样0.05ng/ml、0.1ng/ml、0.2ng/ml、0.5ng/ml(n=6)的空白加标样品,所有重复样品中都能鉴定出分析物的最低浓度作为检测限。通过进样不同浓度的空白基质加标样品,以待测物与内标峰面积的比值为依据考察线性情况,定量下限为标准曲线的最低点。
[0082]4‑
cl
‑
α
‑
pvp、4
‑
mpd和β
‑
th
‑
naphyrone的lod分别为0.1ng/ml、0.05ng/ml、0.1ng/ml;lloq分别为0.5ng/ml、0.2ng/ml、0.2ng/ml。
[0083]
5.3标准曲线、准确度和精密度
[0084]
在两天内进行3个分析批的定量下限及低、中、高质控样品(n=5)分析;通过单一分析批(批内准确度)和不同分析批(批间准确度)获得质控样品值来评价准确度;通过同一分析批(批内精密度)和不同分析批(批间精密度)测量值的相对标准差(变异系数)评价精密度。
[0085]
所有待测物在给定浓度范围内线性良好,其准确度、精密度及线性范围见表2。
[0086]
表2各分析物在不同质控浓度下的精密度、准确度及线性情况
[0087][0088]
所有的精密度rsd(%)均在15%以内,这是内标起到了不可忽视的作用。
[0089]
5.4稀释可靠性、基质效应和回收率
[0090]
将200、2000、20000ng/m的高浓度空白基质加标样品分别用尿液稀释10倍、100倍、1000倍至20ng/ml考察三个稀释因子的可靠性,其准确度误差和精密度rsd均在
±
15%以内。
[0091]
使用6批来自不同供体的空白基质,在低、高浓度下评价基质效应,基质效应结果参见表3,在各质控浓度下,各待测物基质效应cv(%)均小于15%。
[0092]
根据联合国毒品和犯罪问题办事处缉获的材料和生物标本中用于检测非法药物的分析方法验证和设备校准指南在低、中、高浓度下,每个浓度进行5次提取,通过比较提取后加标和加标后提取的分析物峰面积与内标峰面积之比,计算回收率。4
‑
cl
‑
α
‑
pvp、4
‑
mpd和β
‑
th
‑
naphyrone在不同浓度的回收率cv(%)分别为6.99%、9.87%、4.47%。
[0093]
5.5稳定性
[0094]
采用低和髙浓度质控样品,进行下列稳定性考察:
[0095]
短期稳定性:样品在室温下放置7h后处理进样;
[0096]
长期稳定性:样品在
‑
40℃下放置10天后处理进样;
[0097]
冻融稳定性:样品在
‑
40℃下放置1天后在室温下解冻1h,后置于
‑
40℃下如此三个
循环后处理进样;
[0098]
处理过的样品在室温下放置3h及在4℃冰箱中放置14h后进样。
[0099]
基质效应和各稳定性考察结果见表3。
[0100]
表3各分析物在不同质控浓度下的基质效应及稳定性情况
[0101][0102]
由表3可见,各稳定性条件下各待测物的准确度偏差均在
±
15%以内,符合定量要求。
[0103]
6)样品定量
[0104]
取待测尿液样品进行磁分散固相萃取,具体是:向待测尿液样品加内标溶液和缓冲液,离心后取上清液,加入步骤3)所述的疏水性磁珠,混匀2min后入磁珠吸附盘静止1min,去尿液,加纯水震荡清洗2min,再入磁珠吸附盘静止1min,去水加0.1%甲酸
‑
乙腈,洗脱1min,得到含内标物质的待测样品洗脱液;
[0105]
将该含内标物质的待测样品洗脱液进行dart
‑
hrms检测,dart
‑
hrms检测条件同步骤2),获得待测样品与内标物质的峰面积比值,根据步骤4)的标准曲线获得定量结果。
[0106]
实施例2本发明的方法用于大鼠尿液中合成卡西酮类毒品的定量
[0107]
选取sprague dawley雄性大鼠(sprague dawley);体重:270
‑
310g;每个毒品各分配三只大鼠。参考动物实验数据最多的mdpv(horsley rr等,behavioural,pharmacokinetic,metabolic,and hyperthermic profile of 3,4
‑
methylenedioxypyrovalerone(mdpv)in the wistar rat.front psychiatry.2018apr 24;9:144.doi:10.3389/fpsyt.2018.00144.pmid:29740356;pmcid:pmc5928397)及此实验中三种合成卡西酮类毒品的ld
50
,4
‑
cl
‑
α
‑
pvp、4
‑
mpd和β
‑
th
‑
naphyrone分别以8.07mg/kg、5.30mg/kg、1.78mg/kg的剂量皮下注射给药。
[0108]
分别在4h、7h、24h收集大鼠尿液检测原体,24h后每天收集1次至无法检测出原体为止。所有动物实验饲养所需的标准饲料、水、以及动物实验均根据美国国立卫生研究院(2011年修订,第8版)和中国国家医药工业研究院(sopan043,中国上海)发布的《实验动物的护理和使用指南》进行。动物使用许可证号:syxk(shanghai)2019
‑
0027。本发明实验经过上海医药工业研究院实验动物管理和福利、伦理委员会批准。
[0109]
本发明在做动物实验验证方法可行性的同时,研究了其在大鼠尿液中的排泄情况。大鼠尿液中合成卡西酮类毒品的定量结果显示,在0
‑
4小时间,4
‑
cl
‑
α
‑
pvp,4
‑
mpd和β
‑
th
‑
naphyrone在大鼠尿液中的平均浓度分别为223.71ng/ml、934.46ng/ml和26.67ng/ml。4
‑
cl
‑
α
‑
pvp在4
‑
7小时尿液中的含量达到最高,48小时后在尿液中的含量降至纳克级别。相较于其他两个毒品,4
‑
mpd在大鼠尿液中排出量最多,在0
‑
4小时尿液中的含量达到最高,48小时后在尿液中的含量降至纳克级别。β
‑
th
‑
naphyrone在0
‑
4小时尿液中的含量达到最高,
24小时后在所有大鼠尿液中的含量均降至纳克级别,β
‑
th
‑
naphyrone相较于其他两个毒品在大鼠尿液中存在较少。说明本发明的定量方法可以用于尿液中合成卡西酮类毒品的定量研究。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1