一种白前及白前配方颗粒的质量控制方法与流程
1.本发明涉及中药质量控制领域,具体涉及一种白前及白前配方颗粒的质量控制方法。
背景技术:
2.《中国药典》(2020版)规定的萝蘼科植物柳叶白前cynanchum stauntonii(decne.)schltr.ex l
é
vi.或芫花叶白前cynanchum glaucescens(decne.)hand.-mazz.的干燥根茎和根,其功能主治降气,消痰,止咳。白前化学成分复杂,主要含甾体皂苷类、生物碱类、挥发性、黄酮类、氨基酸类等化学成分。
3.甾体皂苷类作为白前的主要特征性和功效性成分,具有较强的抗病毒、抗感染以及抗肿瘤活性。甾体皂苷类成分多经硅胶柱色谱,以高浓度醇及混合有机溶剂洗脱富集得到,水提取物中含量极低。但白前临床用药多采用汤剂的形式,因此白前发挥疗效的基础主要是水溶性成分,并有研究表明,白前水提物具有较强的祛痰、抗炎、抗血栓以及舒张气管等生理活性。故针对白前中水溶性成分进行质量控制,符合临床用药的要求,更能够保证用药的安全性及有效性。
4.目前,未查阅到对白前水溶性成分进行质量控制的质控研究报道。
技术实现要素:
5.因此,本发明提供一种针对白前水溶性成分的整体质量控制方法。
6.一种白前及白前配方颗粒的质量控制方法,包括获取白前的供试品并制备成供试品溶液,采用高效液相色谱法获得供试品溶液的特征图谱,该特征图谱中至少包括尿苷、腺苷和色氨酸所对应的色谱峰。
7.以尿苷所在的特征峰为参照峰s,计算出各个特征峰与参照峰s的相对保留时间,所述特征图谱中还包括相对保留时间在规定值1.4
±
10%以内的特征峰。
8.以尿苷所在的特征峰为参照峰s,计算出各个特征峰与参照峰s的相对保留时间,所述特征图谱中还包括相对保留时间在规定值1.9
±
10%以内的特征峰。
9.其中,所述色氨酸对应的相对保留时间在规定值3.7的
±
10%以内,所述腺苷对应的相对保留时间在规定值4.1的
±
10%以内。
10.所述的高效液相色谱法的色谱条件为:
11.色谱柱:以苯基-己基硅烷键合硅胶为填充剂的色谱柱;检测波长:205nm-280nm;以甲醇为流动相a,以水为流动相b,按以下梯度洗脱程序进行洗脱:
12.13.所述高效液相色谱法中色谱柱的柱温28-32℃,流速0.8-1.2ml/min,理论板数按色氨酸计算应均不低于3000。
14.所述白前的供试品为白前药材、白前饮片、白前水提取物或白前配方颗粒,其中,白前水提取物为白前水提液、白前水提浓缩液、白前水提干燥粉。
15.供试品溶液的制备过程为:取供试品,加入溶剂混合均匀后,密塞,称定重量,预处理,冷却后再称定重量,用溶剂补足减失的重量,摇匀,过滤后获得滤液,滤液即为供试品溶液;
16.所述供试品为白前药材或白前饮片时,预处理的方式为加热回流;所述供试品为白前水提取物或白前配方颗粒时,预处理的方式为超声处理。
17.所述溶剂为质量浓度为0%-100%的甲醇水溶液或0%-100%的乙醇水溶液。即,该溶剂可以为纯水、甲醇、乙醇、甲醇水溶液或乙醇水溶液。
18.所述超声处理的功率为250-300w、频率为40khz,超声处理的时间为15-45min;加热回流的时间为15-45min;优选的溶剂为质量浓度为70%的甲醇。
19.本发明还包括对白前配方颗粒中色氨酸含量的定量控制,包括控制色氨酸在白前配方颗粒中的含量为0.12mg/g~0.65mg/g。
20.所述色氨酸的含量检测也采用高效液相色谱法进行检测,该高效液相色谱法的检测波长为218nm,其他条件与特征图谱相同。
21.本发明技术方案,具有如下优点:
22.1.本发明经过对白前水溶性成分进行深入研究获知,该白前水溶性成分主要包括生物碱类、氨基酸类,其中生物碱类以尿苷、腺苷为主要活性成分,氨基酸类以色氨酸为主要活性成分,基于上述研究发现,本发明提供的质量控制方法,同时实现了尿苷、色氨酸、腺苷各评价指标成分的共同控制,达到白前以及白前配方颗粒的整体质量控制的目的,通过本发明可以对白前配方颗粒的质量判断更加准确、客观,符合临床用药质控需求,保证临床用药的有效性。
23.2.本发明进一步优化获得特征图谱的色谱条件,包括流动相的优化,以及梯度洗脱程序的优化,能够有效改善峰形,提高色谱峰分离度,并且能达到检测高效、灵敏、准确的效果。
24.3.本发明还同时针对色氨酸的含量进行了定量控制,即,控制色氨酸在白前配方颗粒中的含量为0.12mg/g~0.65mg/g;并且,通过本发明的方法还能有效实现物质传递过程的质量控制,具体为:可以有效实现配方颗粒制备过程中中间品(提取液、浓缩液、干燥粉)的质量控制,有效使最终制备得到的白前配方颗粒的成分能够与标准汤剂冻干粉基本一致。
附图说明
25.为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
26.图1是本发明实施例1中白前配方颗粒的特征图谱。
27.图2是本发明实施例1中对照品溶液与供试品溶液的特征图谱。
28.图3是本发明实施例1中白前饮片和白前配方颗粒的特征图谱。
29.图4是本发明实施例2中重复性检测时获得的指纹图谱。
30.图5是本发明实施例4中专属性检测时获得的指纹图谱。
31.图6是本发明实施例5获得的指纹图谱。
32.图7是本发明实施例6获得的指纹图谱。
33.图8是本发明实施例7获得的波长为280nm的指纹图谱。
34.图9是本发明实施例6获得的波长为205nm的指纹图谱。
35.图10是本发明对比例1获得的指纹图谱。
具体实施方式
36.实施例中未注明具体实验步骤或条件者,按照本领域内的文献所描述的常规实验步骤的操作或条件即可进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规试剂产品。
37.仪器:岛津2010aht高效液相色谱仪;uv紫外检测器;岛津20at高效液相色谱仪;pda紫外检测器;me104e电子天平(梅特勒
·
托利多),jy20002电子天平(梅特勒
·
托利多),kq-300db超声波清洗器(昆山市超声仪器有限公司);电子恒温水浴锅dzkw-4(北京中兴伟业仪器有限公司)。
38.试药:尿苷对照品(批号:110887-201803,中国食品药品检定研究院),
39.色氨酸对照品(批号:140686-201904,中国食品药品检定研究院),
40.腺苷对照品(批号:110879-201703,中国食品药品检定研究院),
41.白前对照药材(柳叶白前)(批号:121442-201002,中国药品生物制品检定所),
42.白前配方颗粒:分别取批号为200327-431400-01、200327-431400-02、200327-431400-03、200327-431400-04、200327-431400-06、200327-431400-07、200327-431400-08、200327-438300-09、200327-438300-10、200327-438300-11、200327-438300-12、200327-438300-13、200525-437500-14、200525-437000-15、200608-431400-16的白前饮片经过配方颗粒的制备工艺制备得到的白前配方颗粒。
43.试剂:甲醇(supelco)为色谱纯,甲醇(国药集团化学试剂有限公司)为分析纯,水为屈臣氏纯化水。
44.实施例1
45.一种白前及白前配方颗粒的质量控制方法,包括获取白前的供试品并制备成供试品溶液,采用高效液相色谱法获得供试品溶液的特征图谱,该特征图谱中至少包括尿苷、腺苷和色氨酸所对应的色谱峰。具体的,
46.本实施例中该高效液相色谱法的色谱条件为:以苯基-己基硅烷键合硅胶为填充剂(柱长为250mm,柱内径为4.6mm,粒径为5μm);以甲醇为流动相a,以水为流动相b,按下表1中的规定进行洗脱;流速为每分钟1.0ml;柱温为30℃;检测波长为265nm。理论板数按色氨酸计算应均不低于3000。
47.表1
[0048][0049]
本发明中对照药材参照物溶液的制备过程为:取白前对照药材2.0g,置锥形瓶中,精密加入70%甲醇25ml,密塞,称定重量,超声处理(功率300w,频率40khz)30分钟,取出,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,作为对照药材参照物溶液。另取尿苷、色氨酸、腺苷对照品适量,精密称定,加甲醇制成每1ml分别含10μg的溶液,摇匀,作为对照品参照物溶液。
[0050]
本发明中该供试品为白前配方颗粒时,供试品溶液的制备过程为:取供试品适量,研细,取约0.5g,精密称定,置具塞锥形瓶中,精密加入70%甲醇25ml,密塞,称定重量,超声处理(功率300w,频率40khz)30分钟,取出,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,作为供试品溶液。
[0051]
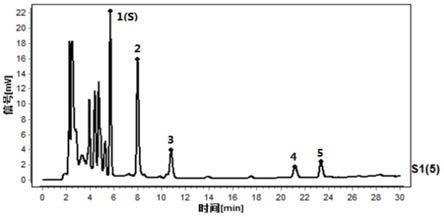
本实施例中采用15批白前饮片制备得到的白前配方颗粒作为供试品进行测定,根据获得的白前配方颗粒的液相图谱使用《中药色谱指纹图谱相似度评价系统》(2012.1版本),以15批液相图谱中s1液相图谱为参照图谱,按中位数计算,得出对照图谱,并进行共有峰的标识,共标识5个共有峰,如图1所示;并获取各供试品对应的液相图谱中各特征峰的保留时间,同时计算出各个特征峰的相对保留时间,如表2所示。
[0052]
表2
[0053][0054][0055]
通过表2可知:各特征峰相对保留时间差异较小,均在
±
10%范围内,符合质量控制要求。
[0056]
并且,采用其中一批供试品溶液与对照品参照物溶液进行检测,检测结果如图2所示,通过对比图2中尿苷、色氨酸、腺苷对照品溶液与供试品溶液中各特征峰,分析确定了峰1为尿苷,峰4为色氨酸,峰5为腺苷。本发明中对照品参照物溶液的制备过程为:分别取尿苷对照品、腺苷对照品和色氨酸对照品适量,精密称定,分别加甲醇制成每1ml含10μg的溶液,作为对照品参照物溶液。
[0057]
同时,采用其中一批的白前饮片、采用该批次白前饮片制备得到的白前配方颗粒进行检测,检测得到的特征图谱如图3所示。其中,白前饮片制备得到供试品溶液的过程为:取白前饮片的粉末约1.0g,精密称定,置具塞锥形瓶中,分别精密加入70%甲醇25ml,密塞,称定重量,加热回流15分钟,取出,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,作为供试品溶液。
[0058]
因此结合特征图谱中各特征峰峰位的情况,在确定峰1、峰4、峰5以对照品色谱峰相对应性进行质控可以有效实现白前质量的评价,确保评价方法的合理性。进一步的,再选择白前对照药材作为随行对照,通过特征峰2、3以1号峰(尿苷)作为s峰计算相对保留时间,结合特征峰2、3的相对保留时间进行质控,能够更有效的从整体性控制白前及白前配方颗粒的质量。
[0059]
实施例2
[0060]
本实施例对特征图谱的重复性、中间精密度和稳定性进行验证,具体如下:
[0061]
1、重复性
[0062]
取相同批次的白前配方颗粒6份,按实施例1中的方法制备供试品溶液并测定,获得如图4所示的特征图谱,其中考察峰1、峰4、峰5与相应的对照品溶液色谱峰对应性,峰2、峰3以峰1为参照峰,计算相对保留时间,并计算rsd,结果如下表3所示:
[0063]
表3
[0064][0065][0066]
小结:根据重复性考察结果,峰1、4、5与相应对照品对应性良好,峰2、3特征峰的相对保留时间rsd在0.0%~0.1%范围内,表明该特征图谱的重复性较好。
[0067]
2、中间精密度
[0068]
采用岛津2010aht、uv紫外检测器,取白前配方颗粒,按实施例1中的方法制备供试品溶液并进行6次测定,获得特征图谱,并以峰1为参照峰,计算相对保留时间,并计算rsd,结果如下表4所示。
[0069]
表4特征图谱中间精密度考察保留时间、相对保留时间
[0070][0071]
小结:采用岛津2010aht、uv紫外检测器获得的特征图谱中,峰1、4、5与相应对照品对应性良好,峰2、3特征峰的相对保留时间rsd在0.1%范围内。不同仪器间峰2、3相对保留时间rsd范围是0.0%~0.9%,结果表明该特征图谱结果在不同仪器间符合分析要求。
[0072]
3、稳定性
[0073]
取白前配方颗粒,按实施例1中的方法制备供试品溶液,分别于0、2、4、8、12、24h按正文方法进行测定,获得特征图谱,分别以峰1为参照峰,计算其相对保留时间,并计算rsd,结果如下表5所示。
[0074]
表5特征图谱稳定性保留时间、相对保留时间
[0075][0076]
小结:由稳定性实验结果可知,各特征峰的相对保留时间rsd在0.5%范围内,表明供试品溶液在24小时内稳定。
[0077]
实施例3
[0078]
本实施例提供的白前配方颗粒的质量控制方法,其除了实施例1中记载的特征图谱的质量控制以外,还包括对白前配方颗粒中质量评价指标成分的定量控制,包括控制色
氨酸在白前配方颗粒中的含量为0.12mg/g~0.65mg/g。
[0079]
本实施例中采用的色谱条件与实施例1基本相同,不同点仅仅在于检测波长调整为218nm后进行检测。
[0080]
采用本实施例的色谱条件,先进行色氨酸标准曲线的绘制,得到求得色氨酸的回归方程为y=26757x-678.37,r=1.0000,其线性范围为1.02-51.23μg/ml。
[0081]
然后采用本实施例的检测方法进行15批次白前饮片、标准汤剂及3批配方颗粒的检测,然后将检测结果代入色氨酸的回归方程计算获得色氨酸含量的百分比含量,计算结果如表6所示。
[0082]
表6
[0083][0084]
通过上述表6可知:白前饮片中色氨酸的含量为0.04mg/g~0.15mg/g,均值为0.10mg/g;白前标准汤剂中色氨酸的含量为0.23mg/g~0.65mg/g,均值为0.44mg/g;三批颗粒中色氨酸的含量为0.14mg/g~0.16mg/g,均值为0.15mg/g,根据生产过程中辅料添加等情况,并考虑到原料的差异情况,确定采用均值的80%作为含量下限选择均值80%为配方颗粒含量限度下限,上限不变(同标准汤剂上限),故确定白前配方颗粒含量为0.12mg/g~0.65mg/g。
[0085]
实施例4
[0086]
本实施例提供了对色氨酸含量检测方法的准确度、重复性、中间精密度、专属性进行验证的过程,具体如下:
[0087]
1准确度
[0088]
取色氨酸对照品适量,精密称定,加70%甲醇溶液制成每1ml含色氨酸12.715μg的溶液。取配方颗粒9份,每份约0.25g,精密称定,每三份分别精密依次加入色氨酸对照品溶液5ml、10ml、15ml,再分别加入70%甲醇各20ml、15ml、10ml,使其70%甲醇加入量为25ml,余下操作同实施例3的操作方法,按照下式计算回收率,结果见表7。
[0089][0090]
表7色氨酸回收率试验结果表
[0091][0092]
小结:回收率试验所测得的色氨酸回收率范围为90.60%~99.30%,平均回收率为94.9%,rsd值为3.8%,符合方法学验证回收率要求,表明利用该方法测得的结果准确。
[0093]
2、重复性
[0094]
取白前配方颗粒6份,按实施例3中的方法制备供试品溶液并测定,检测结果见表8所示。
[0095]
表8色氨酸含量测定重复性试验
[0096][0097]
小结:重复性试验所测得色氨酸平均含量为0.57mg/g,其rsd为0.9%,符合方法学验证重复性要求。
[0098]
3、中间精密度
[0099]
不同分析人员不同时间不同仪器对同一白前配方颗粒进行检测,仪器采用岛津2010aht液相色谱仪(uv紫外检测器)进行中间精密度试验。取配方颗粒6份,按正文方法进行测定,结果如表9所示。
[0100]
表9色氨酸含量测定中间精密度试验
[0101]
[0102]
小结:中间精密度试验所测得色氨酸平均含量为0.56mg/g,rsd值为0.8%,与重复性试验的检测结果的rsd为0.7%,符合方法学验证精密度要求。
[0103]
4、专属性
[0104]
取供试品溶液、色氨酸对照品溶液、阴性对照溶液(70%甲醇溶液)按实施例3中的方法获得其色谱图,如图5所示,通过图5可知,本方法的专属性良好,阴性对照无干扰。
[0105]
5、稳定性
[0106]
取白前配方颗粒,按实施例3中的方法制备供试品溶液,分别于0、2、4、8、12、24h进行测定,记录色氨酸峰面积的变化情况,检测结果见表10。
[0107]
表10
[0108][0109]
由以上数据可知,24小时内色氨酸的峰面积rsd值为2.7%,符合分析要求。
[0110]
实施例5
[0111]
本实施例考察了不同柱温下获得的标准图谱,其他条件与实施例3完全相同,具体的,分别在柱温为28℃、30℃、32℃下进行测定,本实施例获得的指纹图谱如图6所示,检测得到的色氨酸的含量如表11所示。
[0112]
表11
[0113][0114]
不同柱温下测得色氨酸含量rsd值为2.6%,均符合系统适用性要求。表明该方法对不同柱温的耐用性较好。
[0115]
实施例6
[0116]
本实施例考察了不同流速下获得的标准图谱,其他条件与实施例3完全相同,具体的,分别在流速为0.8ml/min、1.0ml/min、1.2ml/min下进行测定,本实施例获得的指纹图谱如图7所示,检测得到的色氨酸的含量如表12所示。
[0117]
表12
[0118][0119]
不同流速下测得色氨酸含量rsd值为1.8%,均符合系统适用性要求。表明该方法
对不同流速的耐用性较好。
[0120]
实施例7
[0121]
本实施例考察了不同波长条件下获得的标准图谱,其他条件与实施例3完全相同,具体的,分别在波长为205nm、280nm下进行测定,本实施例获得的指纹图谱如图8和图9所示,其中,图8为280nm波长条件下的指纹图谱,图9为205nm波长条件下的指纹图谱。
[0122]
对比例1
[0123]
本实施例提供了另一种色谱条件进行质量控制的方法,具体如下:
[0124]
采用与实施例1相同的供试品进行质量控制时,本对比例的色谱条件为:以苯基-己基硅烷键合硅胶为填充剂;以乙腈-水(4:96)为流动相;流速为每分钟0.8ml;柱温为30℃;检测波长为224nm,理论板数按色氨酸计算应均不低于3000。
[0125]
采用上述色谱条件进行检测的得到的检测结果如图10所示,通过图10可知:在该色谱条件下获得的供试品图谱基线不平,且色氨酸、腺苷、尿苷色谱峰峰形不佳,不利于白前配方颗粒的质量控制。
[0126]
显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引伸出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1