一种测定神经生长因子纯度的方法与流程
1.本发明涉及纯度测定技术领域,尤其涉及一种使用反相色谱法测定神经生长因子纯度的方法。
背景技术:
2.神经生长因子(ngf)是神经系统最主要的活性成分之一,它能维持交感神经和感觉神经细胞的生存,具有神经元营养和促突起生长双重生物学功能,对中枢及周围神经元的发育、分化、生长、再生和功能特性的表达均具有重要的调控作用,对保护神经系统的正常功能具有重要作用。
3.鼠神经生长因子的原液中存在不同的分子量异构体,但是现有技术的方法(例如《中国药典》2015版三部中收录的鼠神经生长因子的质量标准中,测定原液的纯度采用的sec
‑ꢀ
hplc法)分辨率不够,无法将不同的分子量异构体分离并进行准确测定,因此无法对原液纯化过程中产生的分子量异构体进行监控,进而难以控制产品质量。
4.有鉴于此,本领域中需要一种能够有效、准确测定神经生长因子纯度的方法。
技术实现要素:
5.本发明的目的是克服现有技术中存在的问题和缺陷,提供一种测定神经生长因子纯度的方法。本发明开发的方法利用反向色谱(例如rp-hplc和/或rp-uplc),并对色谱中的各项条件和参数进行选择和调整后,对神经生长因子原液或原液中ngf纯化过程中的样品进行分离和测定,其基本原理是根据流经色谱柱的各种物质的极性差异将它们分离,极性越大,保留时间越短。由于每种物质在一定条件下具有特定的保留时间,因此可通过色谱图上的保留时间获知目的峰的位置,并通过峰面积归一化法计算ngf原液中ngf纯度,再通过质谱法鉴定出各色谱峰的归属。
6.本发明的方法可对神经生长因子及其分子量异构体进行有效分离,并通过质谱法鉴定出各色谱峰的归属。本发明的方法至少能够有效分离并准确测定神经生长因子的四种剪切形式及主要翻译后修饰型,可对原液的纯化过程进行准确监测,能够精确对产品进行质量控制。
7.定义
8.如本文所使用,术语“色谱法”是指用于化学分离混合物和组分的任何技术,其依赖于对固定相的混合物中的组分的选择性吸引。实例包括吸附色谱法、分配色谱法、离子交换色谱法、尺寸排阻色谱法和亲和色谱法等。
9.如本文所使用,术语“rp-hplc(reversed phase high performance liquidchromatography)”是指反相高效液相色谱法。hplc用于基于化合物的极性以及柱子的固定相来分离化合物。
10.反相色谱法(reversed phase chromatography)是在液相色谱法中使用的洗脱方法,其中流动相的极性显著大于固定相的极性。
11.如本文所使用,术语“rp-uplc(reversed phase ultra performance liquidchromatography)”是指反相超高效液相色谱法。uplc与高效液相色谱法基本相同,不同之处在于:(1)小颗粒、高性能微粒固定相的出现;高效液相色谱的色谱柱,通常使用十八烷基硅胶键合柱,其粒径是5μm,而超高效液相色谱的色谱柱的粒径达到3.5μm,甚至为 1.7μm;(2)超高压输液泵的使用,由于使用的色谱柱粒径减小,使用时所产生的压力也自然成倍增大,故液相色谱的输液泵也相应改变成超高压的输液泵;(3)高速采样速度的灵敏检测器;(4)使用低扩散、低交叉污染自动进样器,配备了针内进样探头和压力辅助进样技术;(5)仪器整体系统优化设计。
12.如本文所使用,术语“流动相”是指引入柱子的溶剂。
13.如本文所使用,术语“固定相”是指在色谱法中使用的、流动相组分对其表现出选择性亲和力的固定相。
14.一方面,本发明提供了一种测定神经生长因子纯度的方法,所述方法使用反相色谱法,色谱条件包括:
15.(1)使用反相色谱柱;
16.(2)流动相:流动相a为含0.06%~0.16%体积比的甲酸水溶液;流动相b为 (0.06%~0.16%)甲酸水溶液-(40~90)乙腈/(10~60)异丙醇(v/v);或流动相b为 (0.06%~0.16%)甲酸水溶液-(80~100)乙腈/(0~20)水(v/v)。在本发明的一个优选实施方式中,流动相a为含0.08%~0.14%体积比的甲酸水溶液;流动相b为(0.08%~0.14%) 甲酸水溶液-(40~90)乙腈/(10~60)异丙醇(v/v);或流动相b为(0.08%~0.14%)甲酸水溶液-(90~100)乙腈/(0~10)水(v/v)。
17.在本发明的一个优选实施方式中,流动相:流动相a为0.09%~0.11%体积比的甲酸水溶液,流动相b为(0.09%~0.11%)甲酸水溶液-50乙腈/50异丙醇(v/v)。
18.更优选地,流动相a为0.1%体积比的甲酸水溶液,流动相b为0.1%甲酸水溶液-50乙腈/50异丙醇(v/v)。
19.在本发明的一个优选实施方式中,所述方法采用梯度洗脱,洗脱液为流动性a和流动相b的混合液,其中,流动相a在混合液中的体积比由78%以上的初始体积比降至66%
‑ꢀ
76%体积比以下完成出峰,再继续下降流动相a的体积比洗脱杂质;其中,流动相中流动相a与流动相b体积比之和为100%。
20.在本发明的一个优选实施方式中,所述反相色谱柱以四烷基硅烷键合硅胶为填充剂。优选地,色谱柱粒度为1.7μm~3.5μm,长度为5cm~15cm,内径为2.1mm~4.6mm;更优选地,所述反相色谱柱为15cm
×
4.6mm,粒度3.5μm。
21.在本发明的一个优选实施方式中,所述反相色谱柱为tsk protein c4-300、agilentadvancebio rp-mab c4、waters protein beh c4。
22.在本发明的一个优选实施方式中,所述反相色谱法为反相高效液相色谱法(rp-hplc) 或反相超高效液相色谱法(rp-uplc)。
23.在本发明的一个优选实施方式中,所述色谱条件还包括:流速为0.1ml/min~1.2ml/min;优选地,所述反相高效液相色谱法的流速为0.2ml/min~1.2ml/min,优选地,所述反相高效液相色谱法的流速为0.2ml/min~0.8ml/min,更优选地,所述反相高效液相色谱法的流速为 0.2ml/min、0.3ml/min、0.4ml/min、0.5ml/min、0.6ml/min、0.7ml/min、
0.8ml/min;所述反相超高效液相色谱法的流速为0.1ml/min~0.5ml/min,优选地,所述反相超高效液相色谱法的流速为0.2ml/min~0.4ml/min;更优选地,所述反相超高效液相色谱法的流速为0.2ml/min、 0.3ml/min、0.4ml/min。
24.在本发明的一个优选实施方式中,所述色谱条件还包括:柱温为25℃~70℃,检测波长为210nm~285nm,进样量为1μg~100μg;优选地,柱温为30℃~45℃,检测波长为 275nm~285nm,进样量为5μg~35μg;更优选地,柱温为35℃,检测波长为280nm,流速为 0.2ml/min,进样量为10μg。
25.在本发明的一个优选实施方式中,所述反相高效液相色谱法(rp-hplc)的梯度洗脱程序如下:
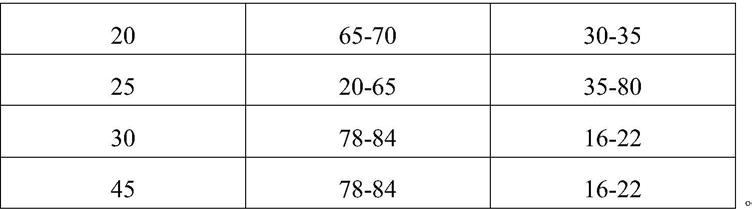
26.时间(分钟)a(体积%)b(体积%)078-8416-222065-7030-352520-6535-803078-8416-224578-8416-22
27.在本发明的一个实施方式中,所述反相超高效液相色谱法(rp-uplc)的梯度洗脱程序如下:
28.时间(分钟)a(体积%)b(体积%)083-8416-1722663425208025.0183-84174083-8417
29.在本发明的具体实施方式中,所述神经生长因子的存在形式包括但不限于:完整型神经生长因子、c端缺失1个氨基酸的神经生长因子、n端缺失8个氨基酸的神经生长因子、 c端缺失1个氨基酸且n端缺失8个氨基酸的神经生长因子,以及神经生长因子糖基化产物和/或神经生长因子氧化产物。
30.另一方面,本发明提供了根据上述方法所得到的神经生长因子,所述神经生长因子的存在形式包括但不限于:完整型神经生长因子、c端缺失1个氨基酸的神经生长因子、n端缺失8个氨基酸的神经生长因子、c端缺失1个氨基酸且n端缺失8个氨基酸的神经生长因子,以及神经生长因子糖基化产物和/或神经生长因子氧化产物。本发明能够测定神经生长因子的纯度。本发明的一些实施方式中,所述神经生长因子为小鼠或人的神经生长因子;优选地,所述神经生长因子为天然小鼠颌下腺或转基因小鼠颌下腺提取的神经生长因子;进一步地,所述转基因小鼠为表达人神经生长因子的转基因小鼠。
31.可以使用本发明的方法检测的样品的来源包括但不限于:神经生长因子原液或原液纯化过程中的样品。在本发明的一些实施方式中,所述神经生长因子为小鼠或人的神经生长因子;优选地,所述神经生长因子为天然小鼠颌下腺或转基因小鼠颌下腺提取的神经生长因子;进一步地,所述转基因小鼠为表达人神经生长因子的转基因小鼠。
32.本发明的方法采用了反向色谱法,对色谱中的各项条件和参数进行选择和调整。
该方法系统适用性好、专属性强、中间精密度好、耐用性强,相较之前的神经生长因子测定方法(例如,sec-hplc法),至少能够有效分离并测定神经生长因子的四种剪切形式及主要翻译后产物,作为原液的纯化过程中的质控指标,能够更好地控制神经生长因子产品的质量。具体地,本发明的方法能够有效分离并测定出包括但不限于以下存在形式的神经生长因子:完整型神经生长因子、c端缺失1个氨基酸的神经生长因子、n端缺失8个氨基酸的神经生长因子、c端缺失1个氨基酸且n端缺失8个氨基酸的神经生长因子,以及神经生长因子糖基化产物和/或神经生长因子氧化产物。
附图说明
33.图1示出了sec-hplc法测定的ngf原液中ngf纯度的色谱图;
34.图2示出了rp-hplc法测定的ngf原液中ngf纯度的色谱图(tsk色谱柱,流动相a:0.1%甲酸-水溶液,b:0.1%甲酸-90%乙腈/10%水溶液);
35.图3示出了rp-hplc法测定的ngf原液中ngf纯度的色谱图(agilent色谱柱,流动相a:0.14%甲酸-水溶液,b:0.14%甲酸-90%乙腈/10%异丙醇);
36.图4示出了rp-hplc法测定的ngf原液中ngf纯度的色谱图(tsk色谱柱,流动相a:0.08%甲酸-水溶液,b:0.08%甲酸-40%乙腈/60%异丙醇);
37.图5示出了rp-hplc法测定的ngf原液中ngf纯度的专属性色谱图(流动相a: 0.1%甲酸-水溶液,b:0.1%甲酸-50%乙腈/50%异丙醇);
38.图6示出了rp-uplc法测定的ngf原液中ngf纯度的色谱图(waters色谱柱,流动相a:0.1%甲酸-水溶液,b:0.1%甲酸-乙腈溶液);
39.图7示出了rp-uplc法测定的ngf原液中ngf纯度的色谱图(agilent色谱柱,流动相a:0.1%甲酸-水溶液,b相:0.1%甲酸-90%乙腈/10%水);
40.图8a和8b示出了不同有机相比例下测定的ngf原液中ngf纯度的色谱图(图8a 的流动相为a:0.1%甲酸-水溶液,b:0.1%甲酸-40%乙腈/60%异丙醇溶液;图8b的流动相为a:0.1%甲酸-水溶液,b:0.1%甲酸-90%乙腈/10%异丙醇溶液);
41.图9a-9b示出了不同柱温下测定的ngf原液中ngf纯度的色谱图。图9a所示结果为柱温30℃,图9b为柱温45℃;
42.图10a-10b示出了不同甲酸浓度下测定的ngf原液中ngf纯度的色谱图。图10a 的流动相为a:0.08%甲酸-水溶液(v/v),b:0.08%甲酸-50%乙腈/50%异丙醇溶液(v/v)),图10b的流动相为a:0.14%甲酸-水溶液(v/v),b:0.14%甲酸-50%乙腈 /50%异丙醇溶液(v/v);
43.图11示出了rp-uplc法测定的ngf原液中ngf纯度的专属性色谱图。
具体实施方式
44.为使本发明要解决的技术问题、采用的技术方案和优点更加清楚,下面将结合附图及具体实施例对本发明进行详细描述。以下实施例用于说明本发明,但不用来限制本发明的范围。
45.下述比较例和实施例中所使用的神经生长因子原液为小鼠颌下腺经专利 cn109762056a实施例5中公开的方法制备获得。
46.下述比较例和实施例中所使用的试剂,如无特别说明,均采用常规方法配制或者由商业途径得到。
47.下述比较例和实施例中所使用的实验方法,如无特别说明,均为常规方法。
48.下述比较例和实施例中所使用的材料、仪器等,如无特别说明,均由商业途径得到。
49.比较例1:sec-hplc法测定ngf原液中ngf纯度
50.一、色谱条件:
51.仪器:shimadzu lc-20ad高效液相色谱仪;
52.凝胶色谱柱:tsk g3000swxl sec,300埃(5μm),7.8
×
300mm;
53.流动相:0.25mol/l磷酸盐缓冲液(含0.15mol/l磷酸氢二钠溶液和0.1mol/l磷酸二氢钠溶液)-乙腈(85∶15,体积比,v/v)
54.流速:0.6ml/min;柱温:30℃;检测波长:280nm;进样量:不低于20μg。
55.二、实验步骤:
56.1、取ngf原液(批号:y20171001,浓度:0.45mg/ml),进样量20μg,注入液相色谱仪,在280nm处检测,记录色谱图,通过色谱图上的保留时间获知目的峰的位置和相关参数,并通过峰面积计算所得到的目标物质的含量。结果见表1、图1。
57.表1 sec-hplc法测定的ngf原液中ngf纯度的结果
58.样品保留时间(min)峰面积纯度ngf原液16.1152140232100%
59.结果显示,sec-hplc法测得的ngf原液中ngf的纯度为100%,但无法分离ngf 及其分子量异构体。
60.实施例1:rp-hplc法测定ngf原液中ngf的纯度
61.一、色谱条件:
62.仪器:waters e2695高效液相色谱仪;
63.色谱柱:tsk protein c4-300 3μm,15cm
×
4.6mm
64.流动相:a:0.1%甲酸-水溶液(体积比,v/v),b:0.1%甲酸-90%乙腈/10%水溶液(v/v)。
65.梯度洗脱程序:
66.时间(min)a(%)(v/v)b(%)(v/v)08020256535302080328020408020
67.柱温:40℃,检测波长:280nm,流速:0.8ml/min,进样量为10μg。
68.二、实验步骤:
69.1、取ngf原液(批号:y20171001,浓度:0.45mg/ml),从中取10μg注入液相色谱仪,在280nm处检测,记录色谱图,采用面积归一化法计算纯度。结果见表2及图2。
70.表2 rp-hplc法测定的ngf原液中ngf纯度的结果
[0071][0072]
由表2及图2的结果可知,本实施例的rp-hplc方法分离出5个已知色谱峰和一个未知峰,峰1~峰5的总纯度为100.00%。通过质谱法鉴定出各色谱峰的归属,峰1~峰5分别为:峰1为ngf糖基化和/或氧化产物,峰2为完整型ngf,峰3为c端缺失一个氨基酸的ngf,峰4为n端缺失8个氨基酸的ngf,峰5为c端缺失一个氨基酸且n端缺失 8个氨基酸的ngf。其中,峰2和峰3的总纯度(%)为57.92%。
[0073]
实施例2:rp-hplc法测定ngf原液中ngf的纯度
[0074]
一、色谱条件:
[0075]
仪器:waters e2695高效液相色谱仪;
[0076]
色谱柱:agilent advancebio rp-mab c4 3.5μm,2.1
×
150mm
[0077]
流动相:a:0.14%甲酸-水溶液(体积比,v/v),b:0.14%甲酸-90%乙腈/10%异丙醇溶液(v/v)。
[0078]
梯度洗脱程序:
[0079]
时间(min)a(%)(v/v)b(%)(v/v)08020107525256832282080308020408020
[0080]
柱温:40℃,检测波长:280nm,流速:0.6ml/min,进样量为10μg。
[0081]
二、实验步骤:
[0082]
1、取ngf原液(批号:y20171001,浓度:0.45mg/ml),从中取10μg注入液相色谱仪,在280nm处检测,记录色谱图,采用面积归一化法计算纯度。结果见表3及图3。
[0083]
表3 rp-hplc法测定的ngf原液中ngf纯度的结果
[0084][0085]
由表3及图3的结果可知,本实施例的rp-hplc法分离出5个已知色谱峰和一个未知峰,峰1~峰5的总纯度为100.00%。通过质谱法鉴定出各色谱峰的归属,峰1~峰5分别为:峰1为ngf糖基化和/或氧化产物,峰2为完整型ngf,峰3为c端缺失一个氨基酸的ngf,峰4为n
端缺失8个氨基酸的ngf,峰5为c端缺失一个氨基酸且n端缺失8 个氨基酸的ngf。其中,峰2和峰3的总纯度(%)为56.70%。
[0086]
实施例3:rp-hplc法测定神经生长因子原液的纯度
[0087]
一、色谱条件:
[0088]
仪器:waters e2695高效液相色谱仪;
[0089]
色谱柱:tskprotein c4-300 3μm,15cm
×
4.6mm
[0090]
流动相:a:0.08%甲酸-水溶液(体积比,v/v),b:0.08%甲酸-40%乙腈/60%异丙醇溶液(v/v)。
[0091]
梯度洗脱程序:
[0092]
时间(min)a(%)(v/v)b(%)(v/v)08416206634252080308416358416
[0093]
柱温:35℃,检测波长:280nm,流速:0.2ml/min,进样量为10μg。
[0094]
二、实验步骤:
[0095]
1、取ngf原液(批号:y20171001,浓度:0.45mg/ml),从中取10μg注入液相色谱仪,在280nm处检测,记录色谱图,采用面积归一化法计算纯度。结果见表4及图4。
[0096]
表4 rp-hplc法测定的ngf原液中ngf纯度的结果
[0097][0098]
由表4及图4的结果可知,本实施例的rp-hplc方法分离出5个已知色谱峰,峰1~峰5的总纯度为100%。通过质谱鉴定出各色谱峰的归属,峰1~峰5分别为:峰1为ngf 糖基化和/或氧化产物,峰2为完整型ngf,峰3为c端缺失一个氨基酸的ngf,峰4为 n端缺失8个氨基酸的ngf,峰5为c端缺失一个氨基酸且n端缺失8个氨基酸的ngf。其中,峰2和峰3的总纯度(%)为57.81%。
[0099]
实施例4:rp-hplc法测定ngf原液中ngf纯度的方法验证
[0100]
一、色谱条件:
[0101]
仪器:waters e2695高效液相色谱仪;
[0102]
色谱柱:tsk proteinc4-300 3μm,15cm
×
4.6mm
[0103]
流动相:a:0.1%甲酸-水溶液(体积比,v/v),b:0.1%甲酸-50%乙腈/50%异丙醇溶液 (v/v)。
[0104]
梯度洗脱程序:
[0105]
时间(min)a(%)(v/v)b(%)(v/v)07822206535252080307822357822
[0106]
柱温:35℃,检测波长:280nm,流速:0.2ml/min,进样量为10μg。
[0107]
二、实验步骤:
[0108]
1、系统适用性
[0109]
采用ngf原液(批号:y20171001,浓度:0.45mg/ml)连续进样6次,出峰顺序为峰1、峰2、峰3、峰4、峰5,各峰保留时间的相对标准偏差(rsd)应不大于1.0%,峰2和峰3百分比之和应不小于50.0%,rsd应不大于2.0%,峰1~峰5的百分比之和应不小于 98.0%,rsd应不大于2.0%;峰3与峰4的分离度应大于1.5,峰2的理论塔板数应不低于 2000。结果见表5。
[0110]
表5系统适用性结果
[0111][0112][0113]
结果表明,ngf原液连续进样6次,峰1~峰5的保留时间rsd均小于1.0%,峰2和峰3百分比之和rsd为0.27%,峰1~峰5的百分比之和rsd为0.00%;峰3与峰4的分离度均大于1.5,峰2的理论塔板数均大于2000。表明该方法的系统适用性良好。
[0114]
2、专属性
[0115]
取空白溶剂缓冲液和y20171001批ngf原液依次进样,记录色谱图,结果见图5。
[0116]
结论:rp-hplc法测定ngf原液中ngf纯度的方法专属性好,可以准确测定各组分的量,可用于ngf原液中ngf的纯度测定。
[0117]
3、中间精密度
[0118]
在不同的检查日期,不同的试验人员,按照确定的色谱条件,对三批ngf原液(批号: y20171001、y20171002、y20171101)进行纯度测定,对结果进行统计,结果见表6。
[0119]
表6中间精密度结果
[0120][0121]
结论:三批ngf原液在不同的检查日期,不同的试验人员进行纯度测定,每批样品的纯度rsd均为0.00,即小于2.0%,说明rp-hplc法测定ngf原液中ngf纯度的中间精密度良好。
[0122]
4、耐用性
[0123]
在确定的色谱条件下,甲酸(fa)比例变化
±
10%、柱温变化
±
2℃、检测波长变化
±
5nm、流速相对值变化
±
10%以及采用三根不同批号的色谱柱对ngf原液(批号:y20171001,浓度:0.45mg/ml)进行纯度测定,结果各条件下测得ngf纯度rsd不大于2.0%,结果见表 7。
[0124]
表7耐用性试验结果
[0125][0126]
结果可知,在甲酸比例、流速、柱温、检测波长发生一定变化以及采用三根不同批号的色谱柱进行原液测定时,对本品纯度的测定无显著差异,各条件下ngf纯度rsd均小于
2.0%,说明本发明的rp-hplc法测定ngf原液中ngf纯度的耐用性良好。
[0127]
实施例5:rp-uplc法测定ngf原液中ngf纯度
[0128]
一、色谱条件:
[0129]
仪器:waters h-class bio超高效液相色谱仪;
[0130]
色谱柱:waters protein beh c4 1.7μm,5cm
×
2.1mm
[0131]
流动相:a:0.1%甲酸-水溶液(v/v),b:0.1%甲酸-乙腈溶液(v/v)
[0132]
梯度洗脱程序:
[0133]
时间(min)a(%)(v/v)b(%)(v/v)08020381191079.520.513772320752523505025.018020308020
[0134]
柱温:30℃,检测波长:280nm,流速:0.2ml/min,进样量为10μl(约5μg)。
[0135]
二、实验步骤:
[0136]
1、取ngf原液(批号:y20171001,浓度:0.45mg/ml),从中取10μg注入液相色谱仪,在280nm处检测,记录色谱图,采用面积归一化法计算纯度。结果见表8及图6。
[0137]
表8 rp-uplc法测定的ngf原液中ngf纯度的结果
[0138][0139]
由表8及图6的结果可知,实施例的rp-uplc法分离出5个色谱峰,峰1~峰5的总纯度为100%。通过质谱法鉴定出各色谱峰的归属,峰1~峰5分别为:峰1为ngf糖基化和/或氧化产物,峰2为完整型ngf,峰3为c端缺失一个氨基酸的ngf,峰4为n端缺失8个氨基酸的ngf,峰5为c端缺失一个氨基酸且n端缺失8个氨基酸的ngf。其中,峰2和峰3的总纯度(%)为60.50%。
[0140]
实施例6:rp-uplc法测定ngf原液中ngf纯度
[0141]
一、色谱条件:
[0142]
仪器:waters h-class bio超高效液相色谱仪;
[0143]
色谱柱:agilent advancebio rp-mab c4 3.5μm,2.1
×
150mm
[0144]
流动相:a:0.1%甲酸-水溶液(v/v),b相:0.1%甲酸-90%乙腈/10%水(v/v)
[0145]
梯度洗脱程序:
[0146]
时间(min)a(%)(v/v)b(%)(v/v)0821814683220208025208025.018218358218
[0147]
柱温:30℃,检测波长:280nm,流速:0.2ml/min,进样量为10μg。
[0148]
二、实验步骤:
[0149]
1、取ngf原液(批号:y20170803,浓度:0.47mg/ml),进样20μg,注入液相色谱仪,在280nm处检测,记录色谱图,采用面积归一化法计算纯度。结果见表9、图7。
[0150]
表9 rp-uplc方法测定的ngf原液中ngf纯度的结果
[0151][0152]
由表9及图7的结果可知,实施例的rp-uplc方法分离出5个色谱峰,峰1~峰5的总纯度为100%。通过质谱鉴定出各色谱峰的归属,峰1~峰5分别为:峰1为ngf糖基化和/或氧化产物,峰2位完整型ngf,峰3为c端缺失一个氨基酸的ngf,峰4为n端缺失8个氨基酸的ngf,峰5为c端缺失一个氨基酸且n端缺失8个氨基酸的ngf。其中,峰2和峰3的总纯度(%)为61.57%。
[0153]
实施例7:rp-uplc法中不同有机相比例测定ngf原液中ngf纯度
[0154]
一、色谱条件:
[0155]
仪器:waters h-class bio超高效液相色谱仪;
[0156]
色谱柱:waters protein beh c4 3.5μm,15cm
×
4.6mm
[0157]
流动相:a:0.1%甲酸-水溶液(v/v),b:0.1%甲酸-40%乙腈/60%异丙醇溶液(v/v) 梯度洗脱程序:
[0158]
时间(min)a(%)(v/v)b(%)(v/v)0841622663425208025.018416358416
[0159]
柱温:35℃,检测波长:280nm,流速:0.2ml/min,进样量为10μg。
[0160]
二、实验步骤:
[0161]
1、取ngf原液(批号:y20170803,浓度:0.47mg/ml),进样20μg,注入液相色谱仪,在
280nm处检测,记录色谱图,采用面积归一化法计算纯度。结果见表10、图8a。
[0162]
表10不同有机相比例测定的ngf原液中ngf纯度结果
[0163][0164]
由表10及图8的结果可知,实施例的rp-uplc方法分离出5个色谱峰,峰1~峰5的总纯度为100%通过质谱鉴定出各色谱峰的归属,峰1~峰5分别为:峰1为ngf糖基化和 /或氧化产物,峰2为完整型ngf,峰3为c端缺失一个氨基酸的ngf,峰4为n端缺失 8个氨基酸的ngf,峰5为c端缺失一个氨基酸且n端缺失8个氨基酸的ngf。其中,峰 2和峰3的总纯度(%)为60.44%。
[0165]
实施例8:rp-uplc法中不同有机相比例测定ngf原液中ngf纯度
[0166]
一、色谱条件:
[0167]
仪器:waters h-class bio超高效液相色谱仪;
[0168]
色谱柱:waters protein beh c4 3.5μm,15cm
×
4.6mm
[0169]
流动相:a:0.1%甲酸-水溶液(v/v),b:0.1%甲酸-90%乙腈/10%异丙醇溶液(v/v)
[0170]
梯度洗脱程序:
[0171][0172][0173]
柱温:35℃,检测波长:280nm,流速:0.2ml/min,进样量为10μg。
[0174]
二、实验步骤:
[0175]
1、取ngf原液(批号:y20170803,浓度:0.47mg/ml),进样20μg,注入液相色谱仪,在280nm处检测,记录色谱图,采用面积归一化法计算纯度。结果见表11、图8b。
[0176]
表11不同有机相比例测定的ngf原液中ngf纯度结果
[0177][0178]
由表11及图8b的结果可知,实施例的rp-uplc方法分离出5个色谱峰,峰1~峰5 的总纯度为100%。通过质谱鉴定出各色谱峰的归属,峰1~峰5分别为:峰1为ngf糖基化和/或氧化产物,峰2为完整型ngf,峰3为c端缺失一个氨基酸的ngf,峰4为n端缺失8个氨基酸的ngf,峰5为c端缺失一个氨基酸且n端缺失8个氨基酸的ngf。其中,峰2和峰3的总纯度(%)为59.82%。
[0179]
实施例9:rp-uplc法中不同柱温测定ngf原液中ngf纯度
[0180]
一、色谱条件:
[0181]
仪器:waters h-class bio超高效液相色谱仪;
[0182]
色谱柱:waters protein beh c4 3.5μm,15cm
×
4.6mm
[0183]
流动相:a:0.1%甲酸-水溶液(v/v),b:0.1%甲酸-50%乙腈/50%异丙醇溶液(v/v)
[0184]
梯度洗脱程序:
[0185][0186][0187]
柱温:30℃,检测波长:280nm,流速:0.2ml/min,进样量为10μg。
[0188]
二、实验步骤:
[0189]
1、取ngf原液(原液由舒泰神(北京)生物制药股份有限公司提供,批号:y20170803,浓度:0.47mg/ml),进样20μg,注入液相色谱仪,在280nm处检测,记录色谱图,采用面积归一化法计算纯度。结果见表12、图9a。
[0190]
表12不同柱温测定的ngf原液中ngf纯度的结果
[0191]
[0192]
由表12及图9a的结果可知,实施例的rp-uplc方法分离出5个色谱峰,峰1~峰5 的总纯度为100%通过质谱鉴定出各色谱峰的归属,峰1~峰5分别为:峰1为ngf糖基化和/或氧化产物,峰2为完整型ngf,峰3为c端缺失一个氨基酸的ngf,峰4为n端缺失8个氨基酸的ngf,峰5为c端缺失一个氨基酸且n端缺失8个氨基酸的ngf。其中,峰2和峰3的总纯度(%)为60.79%。
[0193]
实施例10:rp-uplc法中不同柱温测定ngf原液中ngf纯度
[0194]
一、色谱条件:
[0195]
仪器:waters h-class bio超高效液相色谱仪;
[0196]
色谱柱:waters protein beh c4 3.5μm,15cm
×
4.6mm
[0197]
流动相:a:0.1%甲酸-水溶液(v/v),b:0.1%甲酸-50%乙腈/50%异丙醇溶液(v/v)
[0198]
梯度洗脱程序:
[0199][0200][0201]
柱温:45℃,检测波长:280nm,流速:0.2ml/min,进样量为10μg。
[0202]
二、实验步骤:
[0203]
1、取ngf原液(原液由舒泰神(北京)生物制药股份有限公司提供,批号:y20170803,浓度:0.47mg/ml),进样20μg,注入液相色谱仪,在280nm处检测,记录色谱图,采用面积归一化法计算纯度。结果见表13、图9b。
[0204]
表13不同柱温测定的ngf原液中ngf纯度的结果
[0205][0206]
由表13及图9b的结果可知,实施例的rp-uplc方法分离出5个色谱峰,峰1~峰5 的总纯度为100%。通过质谱鉴定出各色谱峰的归属,峰1~峰5分别为:峰1为ngf糖基化和/或氧化产物,峰2为完整型ngf,峰3为c端缺失一个氨基酸的ngf,峰4为n端缺失8个氨基酸的ngf,峰5为c端缺失一个氨基酸且n端缺失8个氨基酸的ngf。其中,峰2和峰3的总纯度(%)为55.86%。
[0207]
实施例11:rp-uplc法中不同甲酸浓度测定ngf原液中ngf纯度
[0208]
一、色谱条件:
[0209]
仪器:waters h-class bio超高效液相色谱仪;
[0210]
色谱柱:waters protein beh c4 3.5μm,15cm
×
4.6mm
[0211]
流动相:a:0.08%甲酸-水溶液(v/v),b:0.08%甲酸-50%乙腈/50%异丙醇溶液(v/v)
[0212]
梯度洗脱程序:
[0213][0214][0215]
柱温:35℃,检测波长:280nm,流速:0.2ml/min,进样量为10μg。
[0216]
二、实验步骤:
[0217]
1、取ngf原液(原液由舒泰神(北京)生物制药股份有限公司提供,批号:y20170803,浓度:0.47mg/ml),进样20μg,注入液相色谱仪,在280nm处检测,记录色谱图,采用面积归一化法计算纯度。结果见表14、图10a。
[0218]
表14不同甲酸浓度测定的ngf原液中ngf纯度结果
[0219][0220]
由表14及图10a的结果可知,实施例的rp-uplc方法分离出5个色谱峰,峰1~峰5 的总纯度为100%通过质谱鉴定出各色谱峰的归属,峰1~峰5分别为:峰1为ngf糖基化和/或氧化产物,峰2为完整型ngf,峰3为c端缺失一个氨基酸的ngf,峰4为n端缺失8个氨基酸的ngf,峰5为c端缺失一个氨基酸且n端缺失8个氨基酸的ngf。其中,峰2和峰3的总纯度(%)为59.81%。
[0221]
实施例12:rp-uplc法中不同甲酸浓度测定ngf原液中ngf纯度
[0222]
一、色谱条件:
[0223]
仪器:waters h-class bio超高效液相色谱仪;
[0224]
色谱柱:waters protein beh c4 3.5μm,15cm
×
4.6mm
[0225]
流动相:a:0.14%甲酸-水溶液(v/v),b:0.14%甲酸-50%乙腈/50%异丙醇溶液(v/v)
[0226]
梯度洗脱程序:
[0227]
时间(min)a(%)(v/v)b(%)(v/v)083.017.02266.034.02520.080.025.0183.017.03583.017.0
[0228]
柱温:35℃,检测波长:280nm,流速:0.2ml/min,进样量为10μg。
[0229]
二、实验步骤:
[0230]
1、取ngf原液(原液由舒泰神(北京)生物制药股份有限公司提供,批号:y20170803,浓度:0.47mg/ml),进样20μg,注入液相色谱仪,在280nm处检测,记录色谱图,采用面积归一化法计算纯度。结果见表15、图10b。
[0231]
表15不同甲酸浓度测定的ngf原液中ngf纯度的结果
[0232][0233]
由表15及图10b的结果可知,实施例的rp-uplc方法分离出5个色谱峰,峰1~峰5 的总纯度为100%通过质谱鉴定出各色谱峰的归属,峰1~峰5分别为:峰1为ngf糖基化和/或氧化产物,峰2为完整型ngf,峰3为c端缺失一个氨基酸的ngf,峰4为n端缺失8个氨基酸的ngf,峰5为c端缺失一个氨基酸且n端缺失8个氨基酸的ngf。其中,峰2和峰3的总纯度(%)为58.20%。
[0234]
通过实验,对比不同规格、品牌色谱柱、有机相比例、柱温、甲酸浓度,最终摸索出本发明的较佳实施方案,其中所述反相色谱柱选自waters protein beh c4 3.5μm,15cm
×
4.6mm;流动相a为0.1%甲酸-水溶液(v/v),流动相b为0.1%甲酸-50%乙腈/50%异丙醇溶液(v/v),采用梯度洗脱;柱温:35℃,检测波长:280nm,流速:每分钟0.2ml,进样量为10μg。
[0235]
实施例13:rp-uplc法测定ngf原液中ngf纯度的方法验证
[0236]
一、色谱条件:
[0237]
仪器:waters h-class bio超高效液相色谱仪;
[0238]
色谱柱:waters protein beh c4 3.5μm,15cm
×
4.6mm
[0239]
柱温:35℃,检测波长:280nm,流速:0.2ml/min
[0240]
流动相:a:0.1%甲酸-水溶液(v/v),b:0.1%甲酸-50%乙腈/50%异丙醇溶液(v/v)
[0241]
梯度洗脱程序:
[0242]
[0243][0244]
二、实验步骤:
[0245]
1、系统适用性
[0246]
采用ngf原液(批号:y20171002,浓度:0.52mg/ml)连续进样6次,出峰顺序为峰 1、峰2、峰3、峰4、峰5,各峰保留时间的相对标准偏差(rsd)应不大于1.0%,峰2和峰3百分比之和应不小于50.0%,rsd应不大于2.0%,峰1~峰5的百分比之和应不小于 98.0%,rsd应不大于2.0%;峰3与峰4的分离度应大于1.5,峰2的理论塔板数应不低于 2000。结果见表16。
[0247]
表16系统适用性结果
[0248][0249]
结果表明,原液连续进样6次,峰1~峰5的保留时间rsd均小于1.0%,峰2和峰3 百分比之和rsd为0.12%,峰1~峰5的百分比之和rsd为0.00%;峰3与峰4的分离度均大于1.5,峰2的理论塔板数均大于2000。说明rp-uplc法测定ngf原液中ngf纯度的系统适用性良好。
[0250]
2、专属性
[0251]
取空白溶剂缓冲液和y20171002批ngf原液依次进样,记录色谱图,结果见图11。
[0252]
结论:rp-uplc法测定ngf原液中ngf纯度的专属性好,可以准确测定各组分的量,
可用于ngf原液中ngf的纯度测定。
[0253]
3、中间精密度
[0254]
在不同的检查日期,不同的试验人员,按照确定的色谱条件,对三批ngf原液(批号: y20171001、y20171002、y20171101)进行纯度测定,对结果进行统计,结果见表17。
[0255]
表17中间精密度结果表
[0256][0257]
结论:三批原液在不同的检查日期,不同的试验人员进行纯度测定,每批样品的纯度 rsd均小于2.0%,说明rp-uplc法测定ngf原液中ngf纯度的中间精密度良好。
[0258]
4、耐用性
[0259]
在确定的色谱条件下,甲酸(fa)比例变化
±
10%、柱温变化
±
2℃、检测波长变化
±
5nm、流速相对值变化
±
10%以及采用三根不同批号的色谱柱对ngf原液(批号:y20171002,浓度:0.52mg/ml)进行纯度测定,结果各条件下测得ngf纯度rsd不大于2.0%,结果见表 18。
[0260]
表18耐用性试验结果
[0261][0262]
结果可知,在甲酸比例、流速、柱温、检测波长发生一定变化以及采用三根不同批号的色谱柱进行ngf原液测定时,对ngf原液中ngf纯度的测定无显著差异,各条件下 ngf纯度的rsd均小于2.0%,说明本发明的rp-uplc法测定ngf原液中ngf纯度的耐用性良好。
[0263]
综上所述,本发明的方法系统适用性好、专属性强、中间精密度好、耐用性强。
[0264]
以上所述,仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应以所述权利要求的保护范围为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1