匹莫苯丹软咀嚼剂型的杂质检测方法与流程
1.本发明涉及一种匹莫苯丹软咀嚼剂型的杂质的检测方法,属于药物检测技术领域。
背景技术:
2.心脏病是犬猫最常见的老龄病之一,匹莫苯丹是苯并咪唑
‑
哒嗪酮衍生物,是非拟交感神经、非苷类正性肌力药物,已有大量实验证实其治疗犬心力衰竭要优于常规药物,并具有良好的安全性和耐受性,可以明显改善犬的生活质量和生活品质,成为临床应用广泛的治疗心力衰竭药物。患病动物服药的顺应性差,软咀嚼剂型可以大大改善该问题,因此匹莫苯丹软咀嚼剂型是现在研究热点,但软咀嚼剂型基质复杂,且含有较多矫味剂,会对主成分的稳定性存在较大影响。
3.药品在临床使用中的安全性、有效性除了与药品本身的药理活性有关外,也与药品中存在的杂质有很大关系。杂质检测是药品质量控制的一个重要环节,通过药物的杂质检查,可以弄清杂质来源及含量,从而通过优化处方或提高药品包装形式等方式保证药品质量,减少药品不良反应。
4.杂质检测方法应专属性良好,且灵敏度高,对于含有匹莫苯丹的软咀嚼剂型,由于软咀嚼基质的复杂性,且各辅料基本都在紫外处出峰,很难将辅料与杂质区分开,辅料对杂质的检测形成干扰,因此要求杂质检查方法专属性要高,且要尽可能多的检出杂质。
5.欧洲药典对于匹莫苯丹原料药公布的匹莫苯丹杂质的检测方法,由于以上原因无法同时对软咀嚼剂型中的杂质进行检测,尤其是软咀嚼基质的辅料对杂质有明显干扰,造成检测结果不准确,进而影响了药品质量判断的准确性。因此需要设计一种对于软咀嚼剂型专属性良好、能将辅料与杂质区分开、又尽可能多的检出杂质的匹莫苯丹软咀嚼剂型的杂质检测方法。现有技术中,欧洲药典分析方法为:
6.色谱柱:c18(125mm*4.6mm,5μm);
7.流动相a:3g/l磷酸二氢钾(ph2.5);
8.流动相b:乙腈;
9.柱温:45℃;
10.波长:290nm;
11.流速:1.0ml/min;
12.梯度洗脱程序
13.时间(min)流动相a流动相b0
‑
685
→
8015
→
206
‑
2080
→
2020
→
8020
‑
20.120
→
8580
→
1520.1
‑
308515
14.该方法下的杂质分离度如附图1ep方法下软咀嚼中匹莫苯丹杂质检测图谱所示。
15.由图1可知,ep方法下,6
‑
20min处流动相梯度变化较剧烈,基线不平,主峰在该处出峰,辅料干扰主峰及2个已知单杂的出峰,因此该方法并不适用于于软咀嚼剂型下匹莫苯丹杂质的检测。
16.现有技术存在的不足主要在于以下方面:
17.1.现有技术流动相ph均为2.5,在该流动相ph下,软咀嚼剂型中匹莫苯丹杂质与辅料峰不能完全分开,且一般常规色谱柱ph耐受范围为2
‑
8,该流动相ph会大大影响色谱柱使用寿命。
18.2.现有技术为两相梯度洗脱,两相梯度洗脱对于匹莫苯丹软咀嚼的杂质检测来说,并不能将辅料与杂质及主峰分离开来,无法实现杂质的准确检测。
19.3.现有技术条件下,匹莫苯丹主峰出峰时间均在流动相梯度变化剧烈处出峰,流动相梯度变化剧烈会引起基线波动明显,主峰在该处出峰,会影响主峰前后杂质的检出情况,会出现主峰前后的杂质与基线波动无法区分,影响检测灵敏度,影响杂质检测。
20.4.现有技术的流动相体系为磷酸盐
‑
乙腈,而其中磷酸盐主要是钾盐,钾盐对碱性化合物的拖尾改善效果明显,匹莫苯丹虽然是碱性化合物,但由于其溶解性差,在软咀嚼中通常会加入适当的助溶剂或表面活性剂增加其溶解性,而钾盐和某些表面活性剂相溶性差,因此以钾盐做流动相洗脱样品时,会大大影响色谱柱使用寿命。
21.5.对于软咀嚼这种基质复杂的剂型来说,辅料峰通常都有紫外吸收,现有技术均不能完全将辅料峰与杂质及主峰分离开来,进而无法实现杂质检测,这是现有技术最大的缺点。
技术实现要素:
22.本发明目的是提供了一种匹莫苯丹在软咀嚼剂型下的杂质检测方法,以克服现有技术的不足。
23.一种匹莫苯丹软咀嚼剂型的杂质检测方法,其特征在于用三相高效液相色谱进行测定,其中色谱条件如下:
24.色谱柱:c18,4.6mm*250mm、4μm;
25.流动相a:2.6g/l磷酸二氢钠溶液,磷酸调节ph至3.0;
26.流动相b:乙腈
‑
甲醇,80:20;
27.柱温为45℃;
28.检测波长为290nm;
29.流速为1.0ml/min;
30.梯度洗脱:
31.时间(min)流动相a流动相b0
‑
2085
‑
7515
‑
2520
‑
4575
‑
4025
‑
6045
‑
45.0140
‑
8560
‑
1545.01
‑
558515
32.对匹莫苯丹软咀嚼剂型杂质检查供试品溶液在上述液相色谱条件下进行测定,根据主峰及杂质峰判断杂质情况。
33.所述匹莫苯丹软咀嚼剂型的杂质包括杂质a:4
‑
(2
‑
(4
‑
甲氧基苯基)
‑
1h
‑
苯并[d]咪唑
‑5‑
基)
‑3‑
甲基
‑4‑
氧代丁酸,或杂质b:n
‑
(2
‑
氨基
‑4‑
(4
‑
甲基
‑6‑
氧代
‑
1,4,5,6
‑
四氢哒嗪
‑3‑
基)苯基)
‑4‑
甲氧基苯甲酰胺。也包括杂质a、杂质b以外的其他杂质。
[0034]
杂质a:4
‑
(2
‑
(4
‑
甲氧基苯基)
‑
1h
‑
苯并[d]咪唑
‑5‑
基)
‑3‑
甲基
‑4‑
氧代丁酸,匹莫苯丹原料药工艺杂质,结构式如下:
[0035][0036]
杂质b:n
‑
(2
‑
氨基
‑4‑
(4
‑
甲基
‑6‑
氧代
‑
1,4,5,6
‑
四氢哒嗪
‑3‑
基)苯基)
‑4‑
甲氧基苯甲酰胺,匹莫苯丹原料药工艺杂质,结构式如下:
[0037][0038]
用三相高效液相色谱进行测定步骤之前,还包括供试品溶液配制、系统适用性溶液配制与空白辅料溶液配制;
[0039]
所述供试品溶液配制包括:称取研细后匹莫苯丹软咀嚼,置容量瓶中加甲醇超声溶解,稀释至刻度,10000r/min离心10min,取上清液过滤,弃去初滤液,取续滤液作为含匹莫苯丹供试品溶液,且通过甲醇的稀释,使得匹莫苯丹浓度为0.5mg/ml;
[0040]
所述系统适用性溶液配制包括:取杂质a、b及匹莫苯丹对照品适量,精密称取,用甲醇溶解并定量稀释制成每1ml中含杂质a、b各1μg及匹莫苯丹0.5mg的混合对照品溶液,作为系统适用性试验溶液;
[0041]
空白辅料溶液配制包括:按照供试品溶液配制的方式,获得与所述供试品溶液同样体积的空白辅料溶液;
[0042]
将上述各溶液在上述液相色谱条件下进行测定,供试品溶液扣除辅料峰,除杂质a、杂质b外还检出5个未知杂质,主峰前后分离度均符合规定(大于2.0),主峰峰型及理论塔板数也都能满足检测要求(理论塔板数32250),辅料不干扰主峰及各杂质峰检测,说明该方法可以用于匹莫苯丹软咀嚼剂型杂质的检查。
[0043]
除此之外,在该方法下,还能检测出匹莫苯丹软咀嚼剂型中的防腐剂,即可通过本发明专利,同时对制剂中的杂质、防腐剂进行检测。也即该方法在检测匹莫苯丹软咀嚼剂型的防腐剂中的应用。
[0044]
现有技术方法均无法准确测定软咀嚼剂型中匹莫苯丹的杂质,辅料会干扰主峰或者杂质出峰,而在本方法下,可以将辅料与主峰及杂质区分开来,辅料不干扰杂质的检测,且灵敏度高,杂质响应高。
[0045]
本发明优化了色谱柱,将原方法c18(4.6*125mm,5μm)色谱柱优化为c18(4.6*250mm,4μm)色谱柱,更长的柱长以及更小的粒径可以更好地将软咀嚼中的辅料与主峰及杂质峰分离开来。
[0046]
优化流动相体系,将原方法中两相梯度洗脱,优化为三相梯度洗脱,在有机相中加
入甲醇,将水相体系中的磷酸盐由钾盐更换为钠盐,将ph由2.5调整为3.0,该方法可以将匹莫苯丹软咀嚼中辅料与主峰及杂质区分开来,辅料不干扰杂质检测,同时使色谱柱耐用性增加,在一定程度上节省成本。
[0047]
本发明通过调整流动相体系、优化流动相梯度,使软咀嚼剂型下辅料与匹莫苯丹主峰及杂质峰完全分离开,且优化后的流动相体系会大大延长色谱柱使用寿命,增加方法耐用性,优化的流动相梯度使匹莫苯丹主峰在梯度变化缓和处出峰,避免剧烈的梯度变化引起基线波动干扰主峰前后杂质的检测。由此,本发明实现了软咀嚼剂型下匹莫苯丹的杂质检测。
附图说明
[0048]
图1是ep方法下软咀嚼中匹莫苯丹杂质检测图谱。
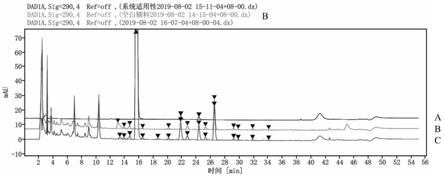
[0049]
图2本方法下匹莫苯丹杂质检测图谱。
[0050]
图3实施例1方法下杂质检测图谱。
[0051]
图4实施例2方法下杂质检测图谱。
[0052]
图5实施例3方法下杂质检测图谱。
[0053]
图6实施例5方法下杂质检测图谱。
具体实施方式
[0054]
本发明使用的各种试剂如下:
[0055]
溶剂:甲醇
[0056]
系统适用性溶液配制:含匹莫苯丹0.5mg/ml,杂质a1μg/ml,杂质b1μg/ml的混合对照品溶液。
[0057]
供试品溶液配制:称取研细后匹莫苯丹软咀嚼剂型适量,得到相当于5mg匹莫苯丹的份量(恋因软咀嚼剂型中的匹莫苯丹均有固定比例,故可通过选取适量的整个匹莫苯丹软咀嚼剂型以获得所需份量的匹莫苯丹),置10ml容量瓶中加甲醇超声溶解,稀释至刻度,10000r/min离心10min,取上清液过滤,弃去初滤液,取续滤液作为含匹莫苯丹0.5mg/ml的供试品溶液。
[0058]
空白辅料溶液配制:称取研细后匹莫苯丹软咀嚼空白辅料适量(约相当于含匹莫苯丹5mg的空白辅料量)置10ml容量瓶中加甲醇超声溶解,稀释至刻度,10000r/min离心10min,取上清液过滤,弃去初滤液,取续滤液作为空白辅料溶液。
[0059]
取上述各溶液照上述色谱条件测定,辅料不干扰主峰及各杂质峰检测。
[0060]
本发明专利方法下匹莫苯丹杂质检测图谱如图2所示,由图2可知,在本发明方法下,主峰出峰处基线平缓,主峰前后分离度良好,辅料峰与主峰及2个已知杂质峰均完全分离,方法专属性强。下面通过多个实施例对本发明做进一步阐述。
[0061]
实施例1
[0062]
在ep方法的基础上调整色谱柱及流动相比例
[0063]
色谱条件:agilent c18色谱柱(4.6*250mm,5μm);流动相a:3g/l磷酸二氢钾溶液(磷酸调节ph至2.5),流动相b:乙腈;检测波长:290nm,流速1.0ml/min;柱温:45℃;
[0064]
时间(min)流动相a流动相b
0.00851510.00802030.00406030.01851540.008515
[0065]
检测结果如图3所示,在实施例1的条件下,辅料在13min、17min分别干扰主峰及杂质检测。
[0066]
实施例2
[0067]
色谱条件:在实施例1的基础上,将流动相a的ph调整为ph3.0,色谱柱为agilent c18(4.6*150mm,4μm)。
[0068]
其检测结果如图4所示,在实施例2的条件下,主峰出峰时间在15min左右,但辅料依然干扰主峰及杂质检测。
[0069]
实施例3
[0070]
通过调整流动相比例观察实验结果
[0071]
色谱条件:agilent c18色谱柱(4.6*150mm,4μm),流动相a:3g/l磷酸二氢钾溶液(磷酸调节ph至3.0),流动相b:乙腈;检测波长:290nm,流速1.0ml/min,柱温:45℃
[0072]
时间(min)流动相a流动相b0.00851510.00802020.00703040.00406040.01851550.008515
[0073]
其检测结果如图5所示,可知在10
‑
20min处的梯度仍需改良。
[0074]
实施例4
[0075]
色谱条件:agilent c18色谱柱(4.6*150mm,4μm),流动相a:3g/l磷酸二氢钾溶液(磷酸调节ph至3.0),流动相b:乙腈,检测波长:290nm,流速1.0ml/min,柱温:45℃;
[0076]
时间(min)流动相a流动相b0.0080205.0080205.01851515.00851525.00703035.00406035.01851545.008020
[0077]
该方法下杂质响应低,主峰峰型差,辅料干扰杂质出峰,可知效果仍不理想。
[0078]
实施例5
[0079]
色谱条件:在实施例3的基础上,将色谱柱调整为agilent c18(4.6*250mm,4μm),
流动相a:2.6g/l磷酸二氢钠溶液(磷酸调节ph至3.0),流动相b:乙腈
‑
甲醇(80:20),检测波长:290nm,流速1.0ml/min,柱温:50℃
[0080]
其检测结果如图6所示,柱温升高至50℃分离度改善不明显,由于柱温升高对色谱柱损耗较大,后续继续以45℃柱温进行方法调整。
[0081]
实施例6
[0082]
色谱条件(本发明方案):agilent c18色谱柱(4.6*250mm,4μm),流动相a:2.6g/l磷酸二氢钠溶液(磷酸调节ph至3.0),流动相b:乙腈
‑
甲醇(80:20),检测波长:290nm,流速1.0ml/min,柱温:45℃;
[0083]
时间(min)流动相a流动相b0.00851520.00752545.00406045.01851555.008515
[0084]
综上,本发明主要提供了一种匹莫苯丹软咀嚼剂型的杂质检测方法,该方法专属性强,灵敏度高,能将复杂的辅料与杂质区分开来,进而很好的控制匹莫苯丹的杂质,为本类制剂的杂质的检查方法提供依据。同时该方法可以延长色谱柱使用寿命,可以节约成本。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1