一种液相咪唑类物质的分析方法及其在环境检测中的应用
1.本发明属于环境分析技术领域,更具体地,涉及一种液相咪唑类物质的分析方法及其在环境检测中的应用。
背景技术:
2.咪唑类物质是一类具有吸光性质的化合物,在大气环境中能够通过羰基化合物与有机胺或铵盐液相反应形成的。由于大气中咪唑类物质可以通过与阳光的相互作用直接影响辐射强迫,也可以通过对云的影响间接影响辐射强迫。咪唑类物质被长距离大气输送后,沉积在雪和冰川的表面,会降低雪的反照率,加速融雪,影响水资源。除了对全球气候影响之外,人体吸入颗粒中咪唑类组成-过渡金属配体化合物能够影响体内氧化应激水平,增加人类的健康风险。所以了解液相反应产物中的咪唑类物质,有利于我们进一步了解咪唑类化合物的大气演化过程和健康风险评估。
3.目前对液相咪唑类化合物的检测技术多数使用液相色谱法,但这类方法的分离效率较低、灵敏度低和基质影响大的缺点。使用气相色谱法对液相咪唑类化合物定性定量,具有分离效率高、灵敏度高、基质影响小等优势。虽然已有关于用双(2-乙基己基)磷酸酯离子对萃取、氯甲酸异丁酯衍生化后通过气相色谱质谱联用仪(gc-ms)对焦糖色素、咖啡等食品中的4-(5-)甲基咪唑进行定性定量分析的报道,但还没有相关报道用于液相咪唑类化合物进行检测。另外,使用双(2-乙基己基)磷酸酯进行离子对萃取,相对于繁琐的固相萃取,这使得样品提纯过程较为简便、快捷。此外,我们建立的新方法既能对小分子咪唑类进行定性定量检测,又可以成功实现分子量较大、检测难度高的分子量较大咪唑类的定性定量检测,具有方法的优越性。
技术实现要素:
4.为了解决上述现有技术存在的不足和缺点,提供一种液相咪唑类物质的分析方法。
5.本发明另一目的在于提供上述所述液相咪唑类物质的分析方法在环境检测中的应用。
6.本发明的目的通过下述技术方案来实现:
7.一种液相咪唑类物质的分析方法,包括以下步骤:
8.s1.将羰基化合物与铵盐溶液混匀反应生成咪唑类物质,加入回收率指示物溶液,制得溶液a;
9.s2.在溶液a或者咪唑类标准物溶液中加入磷酸盐缓冲液,然后再滴加氢氧化钾溶液调节ph至6~9,在室温下涡旋混合均匀后,取出部分溶液后加入与上述取出溶液等体积的双(2-乙基己基)磷酸酯溶液,在室温下涡旋混合均匀后;分为上层为水相,下层为二氯甲烷相,再取二氯甲烷相加入与该溶液等体积的盐酸溶液进行酸化处理,在室温下涡旋混合,得到离子对萃取后的溶液b,溶液b中包括有机相二氯甲烷和水相;
10.s3.取溶液b的水相依次加入乙腈、吡啶、无水乙醇,在室温下涡旋混合,再加入氯甲酸异丁酯,充分反应后加入nahco3溶液和二氯甲烷,涡旋混合后收集下层的有机相,制得咪唑类衍生物;
11.s4.将含有内标物n-甲基甲酰苯胺的二氯甲烷溶液加入步骤s3收集的咪唑类衍生物,转移至顶空瓶中,制得咪唑类衍生物样品溶液;
12.s5.将咪唑类衍生物样品溶液定容后进入气相色谱进行分离,采用质谱检测仪用内标法对咪唑类进行定性定量分析。
13.优选地,步骤s1中所述羰基化合物和铵盐溶液的总体积:回收率指示物溶液的体积比为(200~6000):(1~5);所述羰基化合物和铵盐溶液的体积比为1:1。
14.优选地,步骤s1中所述的羰基化合物为乙二醛或甲基乙二醛,所述铵盐溶液为硫酸铵、甘氨酸或甲胺中一种以上,所述铵盐溶液的浓度为0.01~5mol/l,所述回收率指示物溶液为2-乙基咪唑,所述回收率指示物溶液的浓度为0.001~0.01mol/l。
15.优选地,步骤s2中所述的咪唑类标准物溶液为2,2-联咪唑、咪唑、4-(5-)甲基咪唑、2-乙基咪唑、2,4-二甲基咪唑、2-甲基咪唑-4-甲醛、2-苯基咪唑中的一种以上。
16.优选地,步骤s2中所述磷酸盐缓冲液为磷酸二氢钾和磷酸氢二钾的混合溶液;所述的磷酸盐缓冲液的浓度为0.1~0.3mol/l,所述磷酸盐缓冲液与溶液a或者咪唑类标准物溶液的体积比为(3~10):(2~60)。
17.优选地,步骤s2中所述的双(2-乙基己基)磷酸酯溶液为双(2-乙基己基)磷酸酯的二氯甲烷溶液,其浓度为0.01~0.1mol/l;所述盐酸溶液浓度为0.05~1.2mol/l。
18.优选地,步骤s3所述的移取的水相溶液、乙腈溶液、吡啶溶液、无水乙醇、氯甲酸异丁酯、nahco3溶液、二氯甲烷溶液的体积比为(80~200):(13~25):(6~20):(18~50):(1~10):(80~200):(16~46);所述的nahco3溶液的浓度为0.5~1.14mol/l。
19.优选地,步骤s4中所述的含有内标物n-甲基甲酰苯胺的二氯甲烷溶液的浓度为5~50mg/l。
20.优选地,步骤s5中所述的气相色谱检测条件为:色谱柱为hp-5ms石英毛细管柱;色谱柱升温程序为:70℃,保留1min,20℃/min
→
100℃,保留0min,5℃/min
→
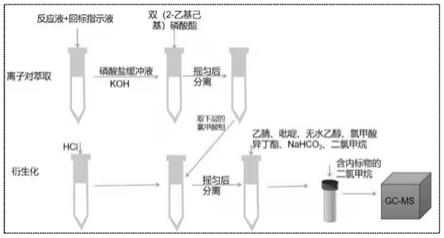
140℃,保留0min,20℃/min
→
280℃,保留3min;进样口温度:250℃,气-质传输线温度:280℃;载气:高纯氦气;流量:1.0ml/min;进样方式:不分流进样;质谱离子源:电子轰击源(ei,70ev);质谱扫描范围:50~550m/z。
21.所述的方法在基于气相色谱法液相样品检测中的应用。
22.与现有技术相比,本发明具有以下有益效果:
23.1.本发明提出的测定液相反应产物咪唑类化合物的分析方法。样品预处理过程简便,衍生化过程快速,基于气相色谱质谱联用法能对液相咪唑类化合物进行很好地定性定量分析。
24.2.本发明可用于液相至少7种以上咪唑类的同时检测,较以往的方法涵盖更多的咪唑类种类,有更广泛的应有前景,能为进一步开展大气液相反应咪唑类化合物的污染特征和来源解析提供技术支持。
25.3.本发明建立了一种快速、简单地实现液相咪唑类组成的分析方法。本发明能应用于大气中液相反应形成产物多种咪唑类物质定性定量的分析,具有操作简单、灵敏度高
和成本低的特点,可以为获取液相反应产物咪唑类定性定量的分析提供技术支持,同时可以应用该方法对大气样品进行定量。
附图说明
26.图1为本发明的测定方法流程简图。
27.图2为实施例1的2,2-联咪唑标准样品色谱图。
28.图3为实施例2的7种咪唑类标准样品色谱图。
29.图4为实施例3的乙二醛与硫酸铵液相反应产物咪唑类色谱图。
具体实施方式
30.下面结合具体实施例进一步说明本发明的内容,但不应理解为对本发明的限制。若未特别指明,实施例中所用的技术手段为本领域技术人员所熟知的常规手段。除非特别说明,本发明采用的试剂、方法和设备为本技术领域常规试剂、方法和设备。
31.实施例1
32.1.取5mg的2,2-联咪唑标样定容至50ml,得100μg/ml的2,2-联咪唑标准溶液。
33.2.取2ml 2,2-联咪唑标准溶液加入2ml的0.2mol/l,ph为6的磷酸盐缓冲液,接着用胶头滴管缓慢滴加浓氢氧化钾溶液调节混合液的ph至8,在室温下涡旋混合30s;混合均匀后,取3ml移至离心管,加入3ml以二氯甲烷为溶剂配置的0.1mol/l双(2-乙基己基)磷酸酯,在室温下涡旋混合30s后以3500r离心2min。分层后,取2ml下层的二氯甲烷相移至第离心管,加入2ml0.1mol/l盐酸酸化,继续在室温下涡旋混合30s,静置1min,得到预处理的溶液。
34.3.将1ml预处理的溶液中的上层水相移至离心管中,依次加入150μl乙腈、100μl吡啶、300μl无水乙醇,在室温下涡旋混合4~5s。接着加入40μl氯甲酸异丁酯溶液,简单晃动后加入1ml 1mol/l的nahco3溶液和250μl二氯甲烷进行萃取,涡旋混合4~5s后收集有机相,制得2,2-联咪唑衍生物。
35.4.将100μl含有浓度为50μg/mln-甲基甲酰苯胺的二氯甲烷溶液加入2,2-联咪唑衍生物中,转移至顶空瓶中,制得2,2-联咪唑衍生物样品溶液。
36.5.将2,2-联咪唑衍生物样品溶液通过自动进样器进入气相色谱进行分离,进入质谱检测仪。色谱条件:色谱柱为hp-5ms石英毛细管柱;色谱柱升温程序为:70℃,保留1min,20℃/min
→
100℃,保留0min,5℃/min
→
140℃,保留0min,20℃/min
→
280℃,保留3min;进样口温度:250℃,气-质传输线温度:280℃;载气:高纯氦气;流量:1.0ml/min;进样方式:不分流进样;质谱离子源:电子轰击源(ei,70ev);质谱扫描范围:50-550m/z。
37.图2为实施例1的2,2-联咪唑标准样品色谱图。从图2中可知,标注is:内标n-甲基甲酰苯胺c8h9no,1:2,2-联咪唑c6h6n4。
38.实施例2
39.1.分别取5mg的咪唑、4-(5-)甲基咪唑、2-乙基咪唑、2,4-二甲基咪唑、2-甲基咪唑-4-甲醛、2-苯基咪唑,2-联咪唑标样定容至50ml,得100μg/ml的咪唑类标准混合溶液。
40.2.取2ml咪唑类标准混合溶液中加入2ml的0.2mol/l,ph为6的磷酸盐缓冲液,接着用胶头滴管缓慢滴加浓氢氧化钾溶液调节混合液的ph至8,在室温下涡旋混合30s。混合均
匀后,取3ml移至第2支离心管,加入3ml以二氯甲烷为溶剂配置的0.1mol/l双(2-乙基己基)磷酸酯,在室温下涡旋混合30s后以3500r离心2min。分层后,取2ml下层的二氯甲烷相移至第3支离心管,加入2ml 0.1mol/l盐酸酸化,继续在室温下涡旋混合30s,得到预处理后的溶液,静置1min。
41.3.将1ml预处理后溶液中的上层水相移至第4支离心管中,依次加入150μl乙腈、100μl吡啶、300μl无水乙醇,在室温下涡旋混合4~5s。接着加入40μl氯甲酸异丁酯溶液,简单晃动后加入1ml 1mol/l的nahco3溶液和250μl二氯甲烷进行萃取,涡旋混合4~5s后收集有机相,制得咪唑类标准品衍生物。
42.4.将100μl含有浓度为50μg/ml n-甲基甲酰苯胺的二氯甲烷溶液加入咪唑类标准品衍生物中,转移至顶空瓶中,制得咪唑类标准品衍生物样品溶液。
43.5.将咪唑类标准品衍生物样品溶液通过自动进样器进入气相色谱进行分离,进入质谱检测仪。色谱条件:色谱柱为hp-5ms石英毛细管柱;色谱柱升温程序为:70℃,保留1min,20℃/min
→
100℃,保留0min,5℃/min
→
140℃,保留0min,20℃/min
→
280℃,保留3min;进样口温度:250℃,气-质传输线温度:280℃;载气:高纯氦气;流量:1.0ml/min;进样方式:不分流进样;质谱离子源:电子轰击源(ei,70ev);质谱扫描范围:50-550m/z。
44.图3为实施例2的7种咪唑类标准样品色谱图。从图3中可知,标注1为咪唑c3h4n2;is为内标n-甲基甲酰苯胺c8h9no,2:4-(5-)甲基咪唑c4h6n2;3:2-乙基咪唑c5h8n2;4:2,4-二甲基咪唑c5h8n2;5:2-甲基咪唑-4-甲醛c5h6n2o;6:2-苯基咪唑c9h8n2;7:2,2-联咪唑c6h6n4。
45.实施例3
46.1.分别配置好0.17mol/l乙二醛和3.3mol/l硫酸铵溶液,以1:1的体积比混匀。取2ml0.17 mol/l乙二醛/3.3mol/l硫酸铵混合液至第1支离心管中,再加入30μl浓度为0.005mol/l的2-乙基咪唑,涡旋混合30s,静置1min。
47.2.往第1支离心管中加入2ml的0.2mol/l,ph为6的磷酸盐缓冲液,接着用胶头滴管缓慢滴加浓氢氧化钾溶液调节混合液的ph至8,在室温下涡旋混合30s。混合均匀后,取3ml移至第2支离心管,加入3ml以二氯甲烷为溶剂配置的0.1mol/l双(2-乙基己基)磷酸酯,在室温下涡旋混合30s后以3500r离心2min。分层后取2ml下层的二氯甲烷相移至第3支离心管,加入2ml 0.1mol/l盐酸酸化,继续在室温下涡旋混合30s,得到预处理后的溶液,静置1min。
48.3.将1ml预处理后溶液中的上层水相移至第4支离心管中,依次加入150μl乙腈、100μl吡啶、300μl无水乙醇,在室温下涡旋混合4~5s。接着加入40μl氯甲酸异丁酯溶液,简单晃动后加入1ml 1mol/l的nahco3溶液和250μl二氯甲烷进行萃取,涡旋混合4~5s后收集有机相,制得咪唑类衍生物。
49.4.将100μl含有浓度为10μg/ml n-甲基甲酰苯胺的二氯甲烷溶液加入咪唑类衍生物中,转移至顶空瓶中,制得咪唑类衍生物样品溶液。
50.5.将咪唑类衍生物样品溶液通过自动进样器进入气相色谱进行分离,进入质谱检测仪。色谱条件:色谱柱为hp-5ms石英毛细管柱;色谱柱升温程序为:70℃,保留1min,20℃/min
→
100℃,保留0min,5℃/min
→
140℃,保留0min,20℃/min
→
280℃,保留3min;进样口温度:250℃,气-质传输线温度:280℃;载气:高纯氦气;流量:1.0ml/min;进样方式:不分流进样;质谱离子源:电子轰击源(ei,70ev);质谱扫描范围:50-550m/z。
51.图4为实施例3的乙二醛与硫酸铵液相反应产物咪唑类色谱图。从图4中可知,标注1:咪唑c3h4n2;is:内标n-甲基甲酰苯胺c8h9no;ss:回收率指示物2-乙基咪唑c5h8n2;2:2,2-联咪唑c6h6n4。说明本发明方法能适用于液相咪唑类组成的定性定量分析。
52.上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合和简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1