一种直接测定谷丙甘氨酸胶囊中3种氨基酸含量的方法与流程
1.本发明涉及一种药物制剂中有效成份的检测方法,特别涉及一种直接测定谷丙甘氨酸胶囊中3种氨基酸含量的方法
背景技术:
2.氨基酸含量检测方法多采用分光光度法、近红外光谱法、高效液相色谱法,其中分光光度法多用于单组份氨基酸或氨基酸总量的测定,且需要用茚三酮衍生化后再测定;近红外光谱法具有高效、无污染、不破坏样品以及同时检测多组分的优点,但存在准确性差的缺点。高效液相色谱法可对多组分氨基酸样品进行测定,测定方法分为柱前衍生法和柱后衍生法。一般采用异硫腈酸苯酯(pitc)、邻苯二甲醛(opa)、2,4-二硝基氟苯(dnfb)等衍生化试剂进行柱前衍生,存在衍生反应条件苛刻(pitc法)、衍生时间长(dnfb法)、衍生物不稳定(opa法)等缺点;柱后衍生法测定氨基酸需先用阳离子交换柱分离氨基酸,再与茚三酮或opa柱后衍生成具有荧光或紫外吸收的物质后测定分析,柱后衍生需要专门的氨基酸分析仪,价格偏贵。
3.谷丙甘氨酸胶囊为复方制剂,其组成成分为谷氨酸,丙氨酸和甘氨酸。主要用于前列腺增生引起的尿频、排尿困难、尿潴留等症的治疗,谷丙甘氨酸胶囊标准收载于《中国药典》2020年版二部,使用dnfb为衍生化试剂,柱前衍生后用hplc法在360nm下测定氨基酸的含量。该方法氨基酸需在碱性条件下与dnfb在60℃水浴中反应50min,衍生化时间较长。氨基柱是由极性基团氨丙硅烷基键合在硅胶上制成的极性键合色谱柱,主要用于分离极性大的物质,如:糖类和氨基酸的分离,待测物质主要因氢键及范德华力的强弱实现分离。本研究采用hplc法,使用氨基柱在210nm波长下直接测定谷丙甘氨酸胶囊中3种氨基酸的含量,避免衍生化法测定氨基酸的一些缺点。
技术实现要素:
4.本发明提供一种直接测定谷丙甘氨酸胶囊中3种氨基酸含量的方法,所述方法,包括以下步骤:
5.1)对照品溶液的制备:称取l-谷氨酸对照品、l-丙氨酸对照品和甘氨酸对照品,置同一量瓶中,加溶液a使溶解,再用乙腈稀释至刻度,作为对照品储备溶液;再经流动相稀释,得到对照品溶液;
6.2)供试品溶液的制备:取谷丙甘氨酸胶囊内容物,置量瓶中,加溶液a使溶解,再用乙腈稀释至刻度;得到供试品溶液;
7.3)测定:将对照品溶液和供试品溶液注入高效液相色谱仪,得到色谱图,根据色谱图计算供试品溶液中谷氨酸,丙氨酸和甘氨酸的含量;
8.其中,所述高效液相色谱仪的色谱条件如下:
9.色谱柱为:sharpsil氨基柱(4.6mm
×
250mm,5μm);
10.流动相:溶液a∶乙腈=43-47∶53-57
11.其中,溶液a为18-22mmol/l磷酸二氢钾溶液,配制方法如下:取磷酸二氢钾2-3g,加水1l溶解后,加浓氨54-6ml,用磷酸调节ph值至4.5-5.5
12.流速:0.8-1.2ml/min;
13.柱温:35-45℃;
14.进样量:10-100μl;
15.检测波长:200-220nm。
16.优选的,所述高效液相色谱仪的色谱条件如下:
17.色谱柱为:sharpsil氨基柱(4.6mm
×
250mm,5μm);
18.流动相:溶液a∶乙腈=45∶55
19.其中,溶液a为20mmol/l磷酸二氢钾溶液,配制方法如下:取磷酸二氢钾2.72g,加水1l溶解后,加浓氨5ml,用磷酸调节ph值至5.0
20.流速:1.0ml/min;
21.柱温:40℃;
22.进样量:50μl;
23.检测波长:210nm;
24.优选的,本发明所述方法,
25.其中,所述对照品溶液的制备,方法如下:精密称取l-谷氨酸对照品90-110mg、l-丙氨酸对照品35-45mg和甘氨酸对照品15-22mg,置同一50ml量瓶中,加溶液a20-30ml,超声2-10min使溶解,再用乙腈稀释至刻度,作为对照品储备溶液;再精密量取上述溶液4-6ml,置10ml量瓶中,加流动相稀释至刻度,摇匀作为对照品溶液。
26.其中,所述供试品溶液的制备,方法如下:取谷丙甘氨酸胶囊内容物混匀,称取70-80mg,置50ml量瓶中,加溶液a20-30ml,超声2-10min使溶解,再用乙腈稀释至刻度,摇匀,作为供试品溶液。
27.最优选的,本发明所述方法,
28.其中,所述对照品溶液的制备,方法如下:精密称取l-谷氨酸对照品102.08mg、l-丙氨酸对照品39.08mg和甘氨酸对照品17.68mg,置同一50ml量瓶中,加溶液a约25ml,超声5min使溶解,再用乙腈稀释至刻度,作为对照品储备溶液;再精密量取上述溶液5ml,置10ml量瓶中,加流动相稀释至刻度,摇匀作为对照品溶液。
29.其中,所述供试品溶液的制备,方法如下:取谷丙甘氨酸胶囊内容物混匀,称取约77mg,置50ml量瓶中,加溶液a约25ml,超声5min使溶解,再用乙腈稀释至刻度,摇匀,作为供试品溶液。
30.其中,所述对照品储备溶液大约含谷氨酸2mg/ml、丙氨酸0.75mg/ml和甘氨酸0.34mg/ml。
31.所述对照品溶液大约含谷氨酸约1mg/ml、丙氨酸0.38mg/ml和甘氨酸0.17mg/ml。
32.所述供试品溶液大约含谷氨酸1mg/ml、丙氨酸0.38mg/ml和甘氨酸0.17mg/ml。本研究建氨基色谱柱hplc法直接测定谷丙甘氨酸胶囊中3种氨基酸含量的方法,通过优化流动相中盐相种类、ph、有机相比例等参数,得到最佳流动相组成。研究证实先用50%溶液a超声溶解样品再用乙腈稀释的方法制备供试品溶液,比只用流动相溶解样品的方法节省15min以上的超声时间,且供试品溶液非常稳定。使用氨基柱采用hplc法在210nm波长下可
直接测定分析上述3种氨基酸,本方法操作简单快捷、经济实用,且线性、精密度和准确度良好,可用于谷丙甘氨酸胶囊中3种氨基酸含量的测定。
33.本发明的优点如下:
34.1、本发明开创性的使用氨基柱测定谷丙甘氨酸胶囊中的3种氨基酸,氨基酸极性非常大,使用c
18
色谱柱对氨基酸几乎无保留,且分离度差。而氨基柱非常适合极性物质分析,采用本发明色谱条件,3种氨基酸能完全分离。
35.2、本发明氨基酸分析无需衍生,取样品按本方法方法稀释后可以直接进样分析,避免衍生化反锁的操作步骤,节省了前处理时间。(谷丙甘氨酸胶囊标准收载于《中国药典》2020年版二部,使用dnfb为衍生化试剂进行柱前衍生后使用hplc法在360nm下测定氨基酸含量。该方法氨基酸需在碱性条件下与dnfb在60℃水浴中反应50min,衍生化时间较长)。
36.3、本发明分析时间短,具有较好的精密度、重复性和准确度。
附图说明:
37.图1:对照品色谱图(1)a.溶液a的色谱图;b.溶液b的色谱图;c.溶液c的色谱图;1.丙氨酸;2.甘氨酸;3.谷氨酸;4.杂质1
38.图2:对照品色谱图(2)a.ph 4.0时的色谱图;b.ph 6.0的色谱图;
39.1.丙氨酸;2.甘氨酸;3.谷氨酸;4.杂质1
40.图3:对照品色谱图(3)a.50%乙腈的色谱图;b.60%乙腈的色谱图;1.丙氨酸;2.甘氨酸;3.谷氨酸;4.杂质1
具体实施方式
41.本发明的上述各项技术特征和在下文(如实施案例)中具体描述的各项技术特征之间都可以互相组合,从而构成新的或优选的技术方案,但本发明不仅仅局限于这些实施例,同样这些实施例也不以任何方式限制本发明。
42.下述实施例中的实验方法,如无特别说明,均为常规方法。下述实施例涉及的制剂若无特别说明,均为普通市售品,皆可通过市场购买获得。
43.下面结合图示和实施例对本发明作进一步详细描述:
44.实施例1:实验条件优化
45.1实验部分
46.1.1仪器、试剂与材料
47.lc-6ad高效液相色谱仪(日本shimadzu公司),ms205du电子天平(瑞士mettler toledo公司),kq-500de数控超声波清洗器(昆山市超声仪器有限公司),fe20k酸度计(瑞士mettler toledo公司)。
48.l-谷氨酸对照品(批号:20210520,含量:98.5%),l-丙氨酸对照品(批号:20210520,含量:98.5%),甘氨酸对照品(批号:20180517,含量:100%),对照品来源均为国药集团化学试剂有限公司。乙腈为色谱纯,其余试剂均为分析纯。
49.谷丙甘氨酸胶囊,生产企业:天津金虹胜利药业有限公司,批号:200903,规格:每粒含谷氨酸0.265g、丙氨酸0.1g、甘氨酸45mg,平均装量为0.410g。
50.1.2色谱条件
51.色谱柱为:sharpsil氨基柱(4.6mm
×
250mm,5μm);
52.流动相:20mmol/l磷酸二氢钾溶液∶乙腈=45∶55
53.其中,20mmol/l磷酸二氢钾溶液,配制方法如下:取磷酸二氢钾2.72g,加水1l溶解后,加浓氨5ml,用磷酸调节ph值至5.0
54.流速:1.0ml/min;
55.柱温:40℃;
56.进样量:50μl;
57.检测波长:210nm。
58.1.3溶液制备
59.对照品溶液的制备:精密称取l-谷氨酸对照品102.08mg、l-丙氨酸对照品39.08mg和甘氨酸对照品17.68mg,置同一50ml量瓶中,加溶液a约25ml,超声5min使溶解,再用乙腈稀释至刻度,作为对照品储备溶液(约含谷氨酸约2mg/ml、丙氨酸0.75mg/ml和甘氨酸0.34mg/ml)。精密量取上述溶液5ml,置10ml量瓶中,加流动相稀释至刻度,摇匀(约含谷氨酸约1mg/ml、丙氨酸0.38mg/ml和甘氨酸0.17mg/ml)。
60.供试品溶液的制备:取谷丙甘氨酸胶囊内容物混匀,称取约77mg,置50ml量瓶中,加溶液a约25ml,超声5min使溶解,再用乙腈稀释至刻度,摇匀(约含谷氨酸1mg/ml、丙氨酸0.38mg/ml和甘氨酸约0.17mg/ml)。
61.2.1流动相缓冲盐种类的选择
62.氨基柱一般作为正相柱使用,在分析氨基酸时分离模式采用亲水作用色谱(hilic)模式,流动相常用乙腈和水。本研究分别以
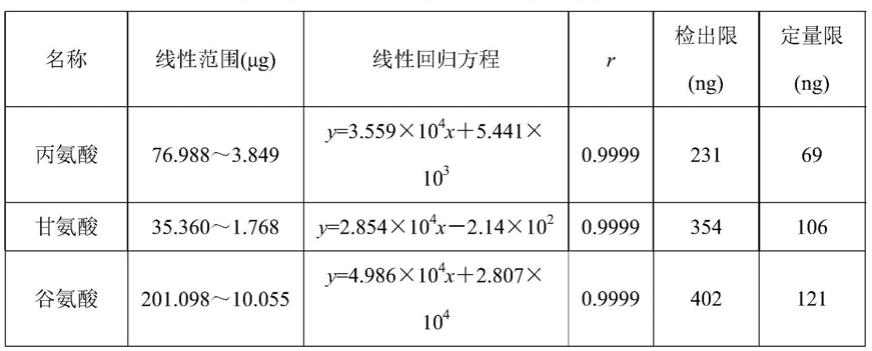
63.溶液a[20mmol/l磷酸二氢钾(氨水调节ph值至5.0)]∶乙腈=45∶55
[0064]
溶液b[20mmol/l磷酸二氢铵(氨水调节ph值至5.0)]∶乙腈=45∶55
[0065]
溶液c[50mmol/l磷酸二氢铵(氨水调节ph值至5.0)]∶乙腈=45∶55考察不同盐相对氨基酸的分离效果。结果采用溶液b时,谷氨酸峰出峰较晚且峰型较差。采用溶液c时,样品和对照品中杂质1峰均与甘氨酸峰重叠。而采用溶液a时,杂质1峰与丙氨酸峰实现基线分离,分离度达到3.5,且谷氨酸峰理论板数高达11000,结果见图1和表1。最终选择溶液a为盐相。
[0066]
表1不同盐相谷氨酸测定结果
[0067]
盐相保留时间(min)理论板数溶液a12.93811174溶液b21.3645984溶液c14.9149627
[0068]
2.2溶液a的ph值优化
[0069]
氨基柱在hilic模式使用时最适ph范围是3~7,在ph范围外会导致色谱柱上的nh2加速水解,色谱柱寿命会下降很快。为延长色谱柱使用寿命,本研究仅选择ph4~6作为流动相ph考察范围。将溶液a分别用磷酸调节ph至4.0、5.0和6.0,考察不同ph时3种氨基酸的分离效果。ph 4.0时,杂质1峰与丙氨酸峰重叠;ph 6.0时,3种氨基酸出峰较快,谷氨酸峰型变差理论板数下降到8600,见图2。ph 5.0时,杂质1峰和丙氨酸峰完全分离,且谷氨酸峰理论板数最高,见图1a。综合优选溶液a用磷酸调节ph值为5.0。
[0070]
2.3乙腈比例的优化
[0071]
调节流动相中乙腈比例为50%、55%和60%,考察不同比例乙腈时3种氨基酸的分离效果。结果乙腈比例为50%时,3种氨基酸出峰较快,且杂质1峰与丙氨酸峰重叠。而乙腈比例为60%时,杂质1峰与丙氨酸峰完全分离,谷氨酸理论板数与55%乙腈时近似,但谷氨酸出峰时间较晚,整体分析时间延长。综合优选流动相中乙腈比例为55%。
[0072]
2.4对照品和供试品溶液制备方法的选择
[0073]
称取对照品和供试品,分别加25ml溶液a和流动相超声溶解,考察两种方法对照品和供试品溶解所需时间。采用溶液a超声溶解时间在5min以内,采用流动相超声溶解时间在20~25min。氨基柱在hilic模式使用时,待测溶液如全部为水溶液,会导致峰型较差,一般加入适量乙腈以减少溶剂效应以改善峰型。最终选择先加50%溶液a超声溶解对照品和供试品后,再用乙腈稀释的方法制备对照品和供试品溶液。
[0074]
2.5方法学考察
[0075]
2.5.1线性关系、定量限与检出限
[0076]
按“色谱条件”,分别量取对照品储备溶液5、10、25、50、75和100μl进样,并记录色谱图。以各氨基酸峰面积为纵坐标(y)、各氨基酸质量(x)为横坐标,进行线性回归。取对照品溶液适量,用流动相稀释成含谷氨酸分别为2.4、3.6、8、12、40μg/ml的对照品浓度,分别按“色谱条件”测定。以各组分信噪比分别满足3∶1作为检出限,满足10∶1作为定量限。线性回归方程、检出限和定量限见表2。3种氨基酸线性方程的相关系数均为0.9999,线性关系良好。检出限在69~121ng之间,定量限在231~402ng之间。
[0077]
表2 3种氨基酸的标准曲线、相关系数、精密度、检出限和定量限
[0078][0079]
2.5.2精密度和稳定性
[0080]
取对照品溶液,按“色谱条件”连续进样6次(50μl),记录色谱图。计算各氨基酸峰面积的rsd,rsd均为0.2%,说明本方法精密度良好。取供试品溶液(称样量77.02mg),在0、2、4、8、24h按“色谱条件”测定,结果3种氨基酸峰面积rsd均小于0.2%,表明3种氨基酸在24h内稳定。
[0081]
2.5.3重复性和样品测定
[0082]
制备6份供试品溶液,按“色谱条件”分析,记录色谱图。以精密度中对照品溶液测定数据以外标法计算样品中氨基酸的含量(按标示量计算),丙氨酸、甘氨酸和丙氨酸的平均含量分别为95.24%、98.89%和96.59%,rsd(n=6)分别为0.9%、1.0%和0.8%,说明本
方法重复性良好。
[0083]
2.5.4准确度
[0084]
称取丙氨酸、甘氨酸和谷氨酸对照品78.56mg、35.75mg和204.2mg,置同一200ml量瓶中,加溶液a约100ml,超声5min使溶解,再用乙腈稀释至刻度,摇匀,作为回收对照品溶液。取谷丙甘氨酸胶囊内容物约308mg,置200ml量瓶中,加溶液a约25ml,超声5min使溶解,再用乙腈稀释至刻度,摇匀,作为回收样品溶液(制备6份)。分别精密量取上述两种溶液各25ml,置50ml量瓶中,摇匀,作为回收测定溶液,按“1.2色谱条件”测定,计算回收率。结果3种氨基酸加样回收率均在98.58%~100.32%之间,且rsd均小于0.6%,说明本方法准确度良好,数据见表3。
[0085]
表3回收率结果表
[0086][0087]
综上所述,本方法操作简单快捷、经济实用,且线性、精密度和准确度良好,可用于谷丙甘氨酸胶囊中3种氨基酸含量的测定。
[0088]
最后需要强调的是,以上所述仅为本发明的优选实施例,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种变化和更改,凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1