一种利用CUT&Tag技术鉴定植物中转录因子与染色质互作的试剂盒及方法
一种利用cut&tag技术鉴定植物中转录因子与染色质互作的试剂盒及方法
技术领域
1.本发明涉及生物技术领域,尤其涉及一种利用cut&tag技术鉴定植物中转录因 子与染色质互作的试剂盒及方法。
背景技术:
2.基因表达调控在多细胞生物的生长和发育中起关键作用。在多细胞生物中,所有 细胞都具有相同的基因组序列。但是,基因组调控,包括dna甲基化、组蛋白修饰、 转录因子及其募集的蛋白质复合物的差异结合,导致不同组织和不同发育时期的基因 表达差异。
3.染色质免疫沉淀(chromatin immunoprecipitation,chip)是一种广泛使用的染色质 分析方法,是用于全基因组dna-蛋白质相互作用的研究的金标准。chip实验的原理 是将目的蛋白和dna的结合用甲醛固定,然后将染色质复合通过机械超声或者酶切 的方式片段化,随后进行基于抗体与靶标蛋白的免疫共沉淀富集作用,从而将与靶标 蛋白互作的染色质片段同时富集下来,进一步地,富集的染色质片段可以用于后续的 高通量测序研究全基因范围内的结合表征,或者用于荧光定量pcr(qpcr)实验验证转 录因子与特殊dna候选位点的结合状况。
4.2019年,西雅图佛瑞德哈金森癌症研究中心开发的cut&tag(cleavage undertargets and tagmentation,cut&tag)技术是一种研究表观基因组染色质分析策略的新 技术。其原理与chip存在本质的差异。从原理上看,cut&tag是一项酶栓系 (enzyme-tethering)策略,它首先利用抗体在完整的细胞或者细胞核内识别靶标蛋白, 随后加入protein a/g融合的tn5转座酶识别与靶标蛋白结合的抗体,因此,tn5转座 酶被栓系在靶标蛋白及其结合的染色质附近,在镁离子的作用下,tn5转座酶对临近 的染色质进行切割并加入dna接头,产生的靶标片段用于后续建库和高通量测序。
5.cut&tag的功能与常规chip技术相同,却有其独特的优势:1)由于原位激活转 座酶而产生的高分辨率和低背景信号;2)因为不需要dna超声处理片段化染色质, 建库过程不需要加接头,大大简化了实验操作和建库流程,节省了实验时间;3)由于 该过程的高灵敏度,因此只需少量起始原料。
6.cut&tag技术是一个全新的技术,仍然在迅速的发展完善过程中。cut&tag技 术最初开发出来的时候被用于动物细胞染色质状态分析。目前cut&tag在组蛋白修 饰研究中的流程/方法已经在动物细胞和植物中都有具体的报道,甚至在动物中,利用 cut&tag进行组蛋白修饰的研究已经发展到了单细胞的水平。然而,由于植物具有 细胞壁组织及多种次生代谢产物的影响,针对特殊植物转录因子与染色质结合研究的 cut&tag流程仍然是个挑战。
7.首先,植物细胞存在细胞壁、大液泡和复杂次生代谢产物,限制了抗体和转座酶 进入到植物细胞内。目前已经有了先提取植物细胞的细胞核,利用细胞核进行 cut&tag反应的植物cut&tag实验的成功报道。然而,目前动植物中只有染色质存 在丰度较高的组蛋
白修饰h3k4me3和h3k27me3被成功鉴定,利用cut&tag鉴定 植物特异的转录因子与dna的结合,目前还未见成功案例报道。这很可能是由于 cut&tag技术主打的可以使用低起始量的细胞的特点造成的。目前已有的纯化和建库 手段也是基于低细胞起始量的cut&tag反应。由于染色质的使用量不够(不如传统 的chip用量多),造成低丰度的转录因子与dna互作的鉴定灵敏度不高。而一旦加 大反应体系中染色质的使用量,过量的未剪切dna又会影响后续的二代建库测序。 因此,如何建立一个适合植物转录因子,特别是低丰度转录因子研究的cut&tag流 程,是个极大的挑战。
8.chip实验与qpcr结合是鉴定转录因子与特定dna区域结合的金标准。常规 cut&tag与传统chip相比存在重要缺陷,即无法进行像chip-qpcr那样的后续荧光 定量pcr实验。这是因为,从原理上看,在cut&tag反应完成之后,未被剪切的(非 靶标)染色质仍然存在于体系中无法去除,无法区分总的input染色质和被片段化的靶 标染色质,从而无法进行后续qpcr实验。这个缺陷极大地限制了cut&tag在鉴定 转录因子特定几个预选的dna结合位点时候的应用。可以想象,研究人员一方面采 用cut&tag结合高通量测序研究在全基因组范围内整体结合特征,却在额外再进行 传统的chip-qpcr实验单独确定转录因子与几个感兴趣的基因位点的结合,这在实验 设计,实验操作和试剂耗材上是及其不科学和不合理的。
9.由于cut&tag的这些不足,目前市面上的基于cut&tag研究染色质状态的试剂 盒均为适合少量样本的试剂盒,也只局限于建库和进行高通量测序的操作,并且只较 好地适合少量动物细胞中的研究。至今,尚无完整的有效的植物转录因子的cut&tag 试剂盒。
技术实现要素:
10.本发明要解决的技术问题是克服现有cut&tag技术在植物中应用的缺陷和不足, 提供一种在动植物、特别是丰度较低的染色质结合蛋白(例如转录因子)与dna的 互作研究中广泛适用的cut&tag-seq和cut&tag-qpcr策略及相应试剂盒,促进表 观遗传调控新技术新手段的发展。
11.具体技术方案如下:
12.本发明提供了一种利用cut&tag技术鉴定植物中转录因子与染色质互作的试剂 盒,其特征在于,包括:生物素化的tn5转座酶、细胞核提取液、洗涤液、抗体杂交 液、链霉亲和素洗涤液、dna洗脱液、一抗、二抗、dna纯化磁珠、链霉亲和素磁 珠、蛋白酶抑制剂、毛地黄皂苷溶液、乙二胺四乙酸溶液、氯化钠溶液、氯化镁溶液、 triton x-100溶液、十二烷基硫酸钠溶液、甘氨酸溶液、预装的phase lock gel凝胶、 dna共沉淀剂和pcr反应液。
13.由于该试剂盒是由含有生物素标记接头序列的转座酶介导的,我们将之称为生物 素化的转座酶介导的cut&tag(biotinylated tn5 transposase mediated cut&tag,b
‑ꢀ
cut&tag)。
14.进一步地,所述生物素化的转座酶由转座酶与生物素化的dna接头引物孵育形 成;所述生物素化的dna接头引物由引物a与引物b退火形成的双链接头i和引物 a与引物c退火形成的双链接头ii构成;引物a包含转座酶识别的mosaic end(me) 序列片段,5’端磷酸化,3'端氨基(aminolinkerc7)修饰;引物b的3’端为与引物 a反向互补的序列,5’端为测序接头序列;引物c的3’端为与引物a反向互补的 序列,5’端为测序接头序列;引物b或引物
c其中的一个的5’端标记有生物素三 甘醇。
15.作为优选,所述tn5转座酶为pg-tn5转座酶或pa-tn5转座酶;细胞核提取液为 含有0.5%-1%体积浓度triton x-100的tris缓冲液;洗涤液为含有蛋白酶抑制剂和毛 地黄皂苷的tris缓冲液;抗体杂交液为含有edta,bsa,蛋白酶抑制剂和毛地黄皂苷 的tris缓冲液;链霉亲和素洗涤液为含edta和tween-20的tris缓冲液;dna洗脱 液为含醋酸钠和甲酰胺的溶液;dna纯化磁珠为表面羧基化修饰的磁珠。
16.作为优选,所述pcr反应液包括:引物i、引物ii和pcr预混液;
17.所述引物i的碱基序列如seq id no.4-seq id no.15所示序列中的一种;所述 引物ii的碱基序列如seq id no.16-seq id no.23所示序列中的一种。
18.本发明涉及一套鉴定植物转录因子与染色质互作的整体解决方法及相关试剂盒。 首先,利用常规的引物修饰的方法,将用于转座酶包埋的接头引物进行生物素标记, 从而在包埋后产生生物素标记的转座子,介导转座子对靶基因位点的切割,同时将自 身携带的生物素化的接头“黏贴”到被切割的dna位点(cut-and-paste原理);其次, 用常规植物dna提取方法获得cut&tag反应总dna后,利用生物素-链霉亲和素结 合的特性,采用链霉亲和素磁珠将生物素化的靶基因dna片段从总dna中纯化出来, 去除未被转录因子和转座酶识别和切割的大量基因组dna;然后,利用聚合酶对结合 在磁珠上的生物素化的双链dna进行磁珠上的延伸反应,将转座酶切割产生的突出 末端补平;接着,利用一定浓度的氢氧化钠在特定温度下对磁珠上结合的双链dna 进行变性,从而将双链dna中没有生物素化的那条单链释放到溶液中,经过进一步 的调节ph到中性、沉淀及清洗等步骤后,得到纯化的、具有测序接头的单链dna, 可用于后续二代测序建库,更重要的,也可以用于荧光定量pcr(qpcr)实验鉴定转 录因子与特定候选位点的结合状态;最后,本发明提供了优化的后续qpcr方案即数据 分析策略。
19.本发明还提供了一种利用cut&tag技术鉴定植物中转录因子与染色质互作的方 法,包括以下步骤:
20.(1)利用生物素化的dna接头引物与tn5转座酶孵育,组装形成生物素化的 tn5转座酶二聚体;
21.(2)固定细胞核内转录因子与染色质的互作状态,提取待测植物组织的细胞核;
22.(3)利用一抗和二抗识别细胞核内与染色质结合的转录因子,再利用生物素化的 转座酶二聚体识别抗体所在区域并切割染色质,形成生物素标记的染色质片段;
23.(4)利用植物基因组dna提取液提取反应后的dna,利用链霉亲和素磁珠对其 中生物素标记的dna片段进行纯化,形成链霉亲和素磁珠-dna片段混合物;
24.(5)以链霉亲和素磁珠-dna片段为模板,进行pcr建库,建立转录因子与染 色质互作的高通量测序文库;
25.(6)文库质检、高通量测序及生物信息学分析;
26.或者,
27.(a)利用生物素化的dna接头引物与tn5转座酶孵育,组装形成生物素化的 tn5转座酶二聚体;
28.(b)固定细胞核内转录因子与染色质的互作状态,提取待测植物组织的细胞核;
29.(c)利用一抗和二抗识别细胞核内与染色质结合的转录因子,再利用生物素化 的
转座酶二聚体识别抗体所在区域并切割染色质,形成生物素标记的染色质片段;
30.(d)利用植物基因组dna提取液提取反应后的dna,取出一部分dna溶液中 dna充作荧光定量pcr的input dna,再利用dna纯化磁珠结合剩余dna中的大 片段,回收上清溶液;
31.(e)利用链霉亲和素磁珠对步骤(d)产物中的小片段进行生物素-亲和素纯化, 形成链霉亲和素磁珠-dna片段混合物;
32.(f)利用dna洗脱液对结合在链霉亲和素磁珠上的dna片段进行洗脱;并以 洗脱下来的dna为模板,进行荧光定量pcr;
33.(g)利用δct方法对荧光定量pcr数据进行分析。
34.进一步地,步骤(1)中,具体步骤包括:
35.a)制备生物素化的dna接头引物:由引物a与引物b退火形成双链接头i,由 引物a与引物c退火形成双链接头ii;
36.引物a包含转座酶识别的mosaic end(me)序列片段,5’端磷酸化,3'端氨基 (aminolinkerc7)修饰;引物b的3’端为与引物a反向互补的序列,5’端为测序 接头序列;引物c的3’端为与引物a反向互补的序列,5’端为测序接头序列;引 物b或引物c中的一个的5’端经生物素三甘醇标记。
37.b)制备tn5转座酶二聚体:将接头i和接头ii以1:1的比例混合形成接头混合物, 接头混合物与tn5转座酶孵育形成tn5转座酶二聚体。
38.作为优选,步骤(5)中,pcr的反应体系包括:引物i、引物ii和pcr预混液; 所述引物i的碱基序列如seq id no.4-seq id no.15所示序列中的一种;所述引物 ii的碱基序列如seq id no.16-seq id no.23所示序列中的一种。pcr的反应程序为: 98℃3min,98℃30s;98℃30s,60℃30s,72℃30s,18-20个循环,72℃5min。
39.作为优选,步骤(d)中,所述dna洗脱液的配方为ph=9.0的含30mm醋酸 钠、95%甲酰胺的溶液;所述dna纯化磁珠为ampure xp beads磁珠。
40.作为优选,步骤(f)中,所述荧光定量pcr的反应体系包括:sybr green,模 板,上游基因特异引物,下游基因特异引物;所述模板分别为步骤(d)中的input dna 和步骤(f)中洗脱下的dna;所述上下游基因特异引物覆盖的目的片段区域包含预 测的转录因子结合基序;反应程序为:98℃3min,98℃30s,60℃30s,72℃30s, 45个循环。
41.进一步地,步骤(g)中,所述δct方法为以下任意一种:
42.(g-1)δct=抗体组样本的ct值-igg对照组样本的ct值,抗体组片段富集倍 数为=2-δc
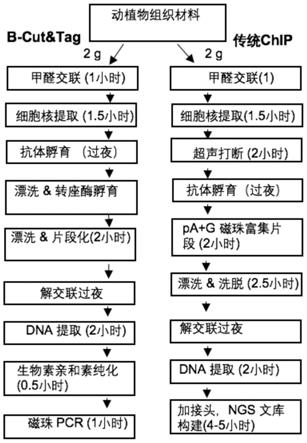
;所述抗体组为使用目的转录因子特异抗体或者抗融合蛋白标签的抗体为一 抗进行cut&tag反应的样本;所述igg对照组指使用igg进行cut&tag反应的样 本;
43.(g-2)δct=cut&tag样本的ct值
–
1%input的ct值,则cut&tag样本中目 的片段占其1%input的比例=2-δct
×
100%。所述cut&tag样本指抗体组或igg对照 组cut&tag。所述1%input dna来自步骤(d);最后,比较目的片段在抗体组和igg 对照组两个比例的差异。
44.经过试验发现,b-cut&tag和高通量测序结合(b-cut&tag-seq)可以应用于高 丰度的组蛋白修饰的研究,其产生的信号与普通的cut&tag信号高度一致。表明 b-cut&tag流程的可行性和准确性。
45.接着,将b-cut&tag和高通量测序(b-cut&tag-seq)应用于普通cut&tag无 法成功
完成的植物转录因子与染色质互作的研究,得到了很好的效果。以植物研究比 较透彻的spl9转录因子为例,利用b-cut&tag进行spl9靶基因的研究,结果表明, spl9靶向与拟南芥植物幼年到成年期转换相关的小rna基因mir172;靶向与开花途 径相关的多个mads box基因(包括ap1,lfy,ful,agl42,soc1等);靶向brc1 基因从而调控植物分枝形成;靶向包括tcl1,try,cpc和etc3在内的myb基因参 与表皮毛的发育;靶向花色素苷和蜡质合成的基因从而参与调控植物次生代谢物的合 成;同时,也结合植物激素赤霉素和甲基茉莉酸信号转导和响应途径的基因,调节激 素响应之间的互作。这些靶基因,与传统染色质免疫共沉淀技术(chip)得到的靶基 因高度吻合,同时也经过启动子、凝胶迁移实验(emsa)等实验进一步确认,具有 高质量的论文研究的支持。因此,b-cut&tag-seq被证明了其产生的结果具备准确性 和稳定性。
46.进一步地,本发明开发了适合b-cut&tag的后续qpcr体系。首先,我们以高 丰度的组蛋白h3k4me3的修饰的qpcr为例,发现与chip-qpcr相同设计策略的引 物对,即基因特异的上下游引物对可以成功用于低染色质input的组蛋白修饰的qpcr 研究,与igg对照组相比,达到很好的富集效果。其产生的富集信号与cut&tag-seq 的信号趋势一致。进一步以植物转录因子atspl9和osphr2为例的研究发现,以高 丰度的组蛋白h3k4me3的修饰的qpcr为例开发的后续qpcr流程同样适用于植物转 录因子的b-cut&tag-qpcr。最后,我们明确了cut&tag-qpcr数据分析的方法, 发现igg对照组的扩增循环数(ct),以及样本自己的未纯化前的1%input,两者均可 以作为计算相对富集倍数或比例的归一化对照。
47.b-cut&tag-seq和b-cut&tag-qpcr体系的发明及在植物中整体实验方案的建 立,有利于在植物中进行转录因子-染色质互作的研究。根据b-cut&tag的原理,大 量未切割的非靶基因的染色质可以通过生物素亲和素纯化去除,不会影响下游的建库 测序。理论上低丰度的转录因子也可以通过灵活调节反应使用的染色质量,或者合并 多次实验染色质进行纯化的策略达成靶基因分析的目的。
48.与现有技术相比,本发明具有以下有益效果:
49.与已有的cut&tag技术相比,b-cut&tag技术通过tn5转座酶将生物素标记的 接头连接到转录因子的靶位点,并通过生物素亲和素纯化步骤消除了未切割的非靶标 染色质,纯化生物素标记的染色质片段,据此原理,可以将反应体系中的染色质用量 根据需求灵活扩大而不影响下游建库测序,该方法是特别为大样本投入量的cut&tag 反应开发的实验流程,在动植物、特别是丰度较低的染色质结合蛋白(例如稀有的组 蛋白修饰,特异的转录因子等)与dna的互作研究中具有很好的应用前景,将极大 地促进生命科学领域基因表达的表观遗传调控的研究。
50.更重要的,与常规的cut&tag技术相比,b-cut&tag及后续qpcr体系的建立, 弥补了常规的cut&tag技术无法进行qpcr的不足,实现了与传统的鉴定转录因子 与染色质特定位点结合的金标准方法,即染色质免疫功沉淀结合qpcr(chip-qpcr) 同等的功能,并且在实验操作上比chip更简洁。
附图说明
51.图1为本发明cut&tag试剂盒的技术原理。
52.图2为b-cut&tag工作流程与传统染色质免疫共沉淀(chip)的工作流程比较。
53.图3为应用b-cut&tag技术进行拟南芥植物叶片细胞组蛋白h3k4me3修饰状态 分析结果;
54.其中,a为不同样本之间测序信号分布的相关性分析结果;b为三个不同cut&tag 样本的信号在编码基因附近的热图。
55.图4为应用b-cut&tag进行spl9的靶基因研究实验过程电泳胶图;
56.其中,两个anti-flag样本(anti-flag rep1和anti-flag rep2)为两个实验重复, 两个igg样本为两个负对照;样本为经过链霉亲和素纯化后未结合的dna。
57.图5为应用b-cut&tag-seq进行spl9的靶基因的分析产生信号的分布热图;其 中,两个spl9为两个实验重复,两个igg样本为两个负对照。
58.图6为应用b-cut&tag技术成功鉴定的一些已知的spl9的靶基因的总结图。
59.图7为应用b-cut&tag-qpcr对特定gb_d10g1774基因位点两个不同的区域 的h3k4me3组蛋白修饰水平进行鉴定的结果图。
60.其中,a为tn5转座酶切割标靶染色质的四种粘贴接头的组合的示意图;b为以基 因gb_d10g1774为例子,从上到下分别为gb_d10g1774位点cut&tag-seq结果的igv 图,比对到不同区域的reads的个数分别在h3k4me3实验组与igg对照组的统计图, 以及不同区域的片段在h3k4me3实验组与igg对照组的相对含量的cut&tag-qpcr 结果图;c为相同的基因位点相应区域的qpcr结果,其中对照组为是样本自身生物 素亲和素纯化之前的dna,即input dna。
61.图8展示了b-cut&tag-qpcr中使用引物p1造成非特异性扩增。
62.其中,a为qpcr中的p1单引物非特异性扩增造成原因的示意图;b为实验组和 对照组在p1单引物存在的情况下qpcr扩增曲线图。
63.图9为应用b-cut&tag-qpcr鉴定拟南芥spl9转录因子和水稻phr2转录因 子与其靶基因的结合结果图。
64.其中,a为拟南芥spl9转录因子b-cut&tag-seq结果中三个靶基因位点的信号 富集情况的igv图,同时atact2基因作为对照;b图为b-cut&tag-seq的数据中比 对到qpcr目标区域(a图标记)的rpm(每百万reads中的比对到该区域的reads数)在 rspl9实验组与igg对照组的比较图;c图为rspl9实验组与igg对照组相比时, attcl1、attry、atful和atact2目标区域富集程度的b-cut&tag-qpcr结果图;d 图为水稻phr2转录因子已知的三个靶基因启动子区域p1bs基序的分布图;e图为在 磷元素缺乏(-p)和充足(+p)不同的处理条件下,phr2实验组与igg对照组相比时, ospt2、ospt8、osram1目标区域富集程度的b-cut&tag-qpcr结果图。
具体实施方式
65.以下结合说明书附图附表和具体实施例来进一步说明本发明,但实施例并不对本 发明做任何形式的限定。除非特别说明,本发明采用的试剂、方法和设备为本技术领 域常规试剂、方法和设备。除非特别说明,以下实施例所用试剂和材料均为市购试剂 配置。
66.实施例1b-cut&tag技术原理和操作流程
67.一、带生物素标记接头的tn5转座酶的生成
68.实验中使用的蛋白a融合的tn5转座酶为市购,浓度为(500ng/μl or 7.5pmol/μ
l)。
69.1.1接头引物合成:
70.使用的引物接头通过常规引物和修饰引物合成获得,三条引物,包括引物a,引 物b和引物c;其碱基序列依次如seq id no.1,seq id no.2和seq id no.3所示。 其中,引物a为转座酶识别的me序列片段,5'-phosphate,3'-aminolinkerc7修饰;引 物b的3’端为与引物a反向互补的序列,5’端为测序接头序列5’端biotin teg修饰; 引物c为普通的引物,其3’端为与引物a反向互补的序列,5’端为测序接头序列。
71.不同的测序平台接头序列有所不同,并不局限于此序列。引物具体序列见表1。
72.表1接头引物列表
[0073][0074]
1.2引物退火形成双链接头:
[0075]
按照1od每管分装的引物。加入退火缓冲液将引物分配置成100μm的母液(可 以长期储存在-20度),然后在两个pcr管中设置如下反应:
[0076]
反应1(接头ab),10μl引物a(100μm),10μl 100μm引物b(100μm);
[0077]
反应2(接头ac),10μl引物a(100μm),10μl 100μm引物c(100μm)。
[0078]
以上管子置于pcr仪中,运行以下程序:使用热盖,75℃,15分钟;60℃, 10分钟;50℃,10分钟;40℃,10分钟;25℃,30分钟)。最终得到的接头 的浓度为每重接头50pmol/μl。
[0079]
1.3生物素化转座酶生成(形成转座酶二聚体):
[0080]
在1.5ml离心管中设置以下反应进行转座酶的包埋:10μl pa-tn5转座酶(500 ng/μl or 7.5pmol/μl),0.75μl接头ab(50pmol/μl),0.75μl接头ac(50pmol/μl), 7.25μl包埋缓冲液,总共18.75μl。因此,体系中接头混合物与转座酶的比例为1:1。 用枪头轻柔吹打20次混合均匀,置于30度水浴锅中1小时,生成的转座酶浓度为4 pmol/μl,包埋好的转座酶保存在-20度。
[0081]
二、试剂盒的各组分配置
[0082]
试剂盒的各组分配置方案(以最优组成为例),如下:
[0083]
包括1个核心酶(pa-tn5或者pg-tn5转座酶),5个缓冲液贮存液(10
×
isob, 10
×
wb,5
×
ab,2
×
sa wb,eb),2种磁珠,2个抗体(igg负对照抗体和h3k4me3 正对照抗体,另外还有11个其他试剂/耗材,配置成表2所示的浓度。其中,5个缓冲 液贮存液按照表3的配方配置,在实验开始前,按照表4提供的配方配成工作液,现 配现用。
[0084]
表2试剂盒组分列表
[0085][0086][0087]
表3储存液配方
[0088][0089][0090]
表4工作液配方(现配现用)
提取待测植物组织的完整细胞核;
[0096]
其中,提取关键为裂解后的细胞经过500目的滤网过滤,收集通过滤网的细胞核, 并用wb(c+)缓冲液轻柔洗涤;wb(c+)缓冲液为试剂盒提供的10x wb母液稀释到1x 浓度后,以1:1000加入蛋白酶抑制剂cocktail。
[0097]
(b)生成带生物素标记接头的tn5转座酶(见第一部分)。
[0098]
(c)一抗孵育:将细胞核重悬在ab缓冲液中,加入一抗进行孵育;
[0099]
其中,细胞核体积(300
×
g-400
×
g离心力下的体积)与ab缓冲液的体积比为 1:20,即50μl细胞核重悬在1ml ab(缓冲液;每个反应上述细胞核悬液的用量为 250μl,一抗的含量为2μg(1:125稀释);ab缓冲液为试剂盒提供的5xab母液稀释 到1x浓度后以1:1000加入蛋白酶抑制剂cocktail,以1:50(0.1%终浓度)或1:100(0.05% 终浓度)体积比加入5%w/v毛地黄皂苷(digitonin)。
[0100]
(d)二抗孵育:将细胞核温柔洗涤除去不结合的一抗,加入二抗进行信号的放大; 其中,二抗的含量为250μl wb(c+d+)缓冲液含1μg(1:250稀释),wb(c+d+)缓 冲液为试剂盒提供的10x wb母液稀释到1x浓度后,以1:1000加入蛋白酶抑制剂 cocktail,以1:100(0.05%终浓度)体积比加入5%w/v毛地黄皂苷(digitonin)。
[0101]
(e)转座酶孵育:将细胞核温柔洗涤除去不结合的二抗,加入pa-tn5转座酶孵 育;其中,tn5转座酶的含量为250μl t i b缓冲液含8pmol pa-tn5转座酶,t i b 缓冲液为步骤(h)中wb(c+d+)以1:20体积比加入3m氯化钠溶液。
[0102]
(f)片段化:将细胞核温柔洗涤除去不结合的pa-tn5转座酶,加入tb缓冲液 进行片段化反应。其中,tb缓冲液为步骤(h)中wb(c+d+)以1:20体积比加入3m氯 化钠溶液,以1:100体积比加入1m氯化镁溶液,并在37℃下切割1小时。
[0103]
(g)dna的生物素亲和素纯化:步骤(f)反应结束后,按照常规的方法提取 dna,并进行链霉亲和素磁珠纯化;将dna加入预清洗后的链霉亲和素磁珠中,在 室温下旋转孵育30~40分钟,将生物素化的dna片段结合到链霉亲和素磁珠上,清 洗后,得到与链霉亲和素磁珠-dna混合物,最后重悬在无菌水中;
[0104]
其中,链霉亲和素磁珠的浓度为10mg/ml,其中每毫克磁珠能结合500-3500pmol 生物素化的dna片段;所述链霉亲和素磁珠的用量为5-10μl;链霉亲和素磁珠预清 洗活化的方法为:用含有1
×
sa wb缓冲液洗涤链霉亲和素磁珠2次,再用2
×
sa wb 缓冲液洗1次,最后重悬于2
×
sa wb缓冲液中;2
×
sa wb缓冲液为试剂盒提供, 1
×
sa wb由2
×
sa wb缓冲液稀释可得;磁珠-dna混合物最后重悬在50μl无菌水 中。
[0105]
(h)磁珠上pcr:以步骤(g)的产物为模版进行pcr建库,反应体系为:步骤 (g)的产物(42μl),4μl引物n70x,4μl引物n50x,50μl pcr mix;反应程序 为:98℃3min,98℃30s;98℃30s,60℃30s,72℃30s,18-20个循环,72℃5 min;其中,引物n701-n712的碱基序列如seq id no.4-seq id no.15所示;所述 引物n501-n508的碱基序列如seq id no.16-seq id no.23所示。
[0106]
(i)文库质检、高通量测序及生物信息学分析。
[0107]
b-cut&tag技术原理如图1展示,利用生物素标记的转座酶dna接头,在转座 酶切割染色质时将生物素标记的引物接头加入到dna片段中,通过后续的生物素-链 霉亲和素系统纯化dna,然后进行pcr建库。
[0108]
与常规cut&tag相比,b-cut&tag接头引物是生物素标记的;因此包埋结束后, 成熟的转座子被加入了生物素标记的接头。在细胞核内,与一段染色质结合的目标蛋 白被其特异抗体或者被目的蛋白融合标签的抗体识别,随后蛋白a融合的tn5转座酶 识别并结合抗体,在镁离子的激活下,转座酶激活并在原位切割染色质。tn5转座酶 的工作模式为切割并粘贴,将染色质切割后,把接头序列加入到被切割的位点,由于 b-cut&tag中接头是带生物素标记的,由此产生的染色质片度最终被生物素标记。
[0109]
b-cut&tag与普通cut&tag第二个最大的差别是后续的生物素亲和素纯化和磁 珠原位pcr建库。片段化的染色质由于带有生物素标记,可以利用亲和素磁珠进行纯 化。由于生物素-亲和素的结合是非常稳定的配体和受体的结合,一旦发生结合,就很 难将两者分开。因此,b-cut&tag开发了磁珠原位pcr建库。由于只标记了一条接 头引物,tn5转座酶片段化产生的双链dna其中有一条链没有被生物素标记,可以 在pcr加热程序下dna变性,核酸双螺旋结构中碱基对的氢键断裂,使得双链变为 单链的过程作为扩增的模版,从而极大程度降低磁珠对pcr的影响。
[0110]
b-cut&tag技术与传统的chip相比,仍具备不可比拟的优势。图2展示了 b-cut&tag与chip流程的比较。可以看出,b-cut&tag不需要超声打断染色质,而 超声打断染色质的好坏往往是直接决定chip反应能否成功。对于不同的固定时间不同 量的染色质,进行超声打断的条件摸索是一个极具挑战的过程,后续的解交联验证打 断效果也需要耗费大量时间。此外,b-cut&tag技术在片段化的过程中直接加入了测 序的接头引物,后续可以直接进行pcr建库,而chip后续的建库需要相应的试剂盒 进行加接头后进行建库,从时间成本、试剂成本以及操作的简洁性看,b-cut&tag 和cut&tag一样,仍具备很大优势。
[0111]
实施例2应用b-cut&tag-seq进行染色质组蛋白修饰的研究
[0112]
本实施例首先对b-cut&tag技术的可行性与信号的稳定性进行了研究。采用组 蛋白修饰h3k4me3为研究例子,按照文献报道的针对组蛋白的小量input染色质的实 验体系(参考文献doihttps://doi.org/10.1186/s13007-020-00664-8),比较了 b-cut&tag-seq与常规cut&tag-seq在信号上的一致性和稳定性。本实施例还设置 了一个b-cut&tag-seq不进行链霉亲和素磁珠纯化而是和常规cut&tag一样直接将 提取的dna进行pcr的样本(b-cut&tag beads-),用来检测生物素接头标记的转座 酶活性是否受到影响。此外,本实施例还设置了igg的负对照,用于评估背景剪切的 强弱。
[0113]
结果表明,h3k4me3的cut&tag,b-cut&tag beads-和b-cut&tag三个样本, 从信号的一致性看,三个样本之间信号具备高度的一致性(相关系数》0.99)(图3a)。 用macs2软件进行富集的峰的分析,分别从cut&tag,b-cut&tag beads-和 b-cut&tag三个样本中得到17076,18632和17309个峰,数目相近,且它们在基因 附近的信号强度和特征高度一致(图3b)。
[0114]
以上结果表明,b-cut&tag从技术上具备可行性,生物素标记和磁珠原位pcr 并不会对信号造成影响,即产生的信号与常规cut&tag高度一致。这是进一步进行 转录因子b-cut&tag的基础。
[0115]
实施例3应用b-cut&tag-seq进行转录因子-dna互作的研究
[0116]
本实施例将b-cut&tag与二代测序结合(b-cut&tag-seq)应用于植物转录因 子与dna互作的研究,用于鉴定转录因子的靶基因位点。本实施例择了在植物中研 究较多的转
录因子spl9作为例子,探索b-cut&tag是否可以用于植物转录因子靶基 因的研究。
[0117]
spl9被报道参与了多种发育和代谢途径的调控,包括幼年期到成年期的转变, 开花,表皮毛的发育,花色素苷和表皮蜡质的合成,分枝发育,通过靶向甲基茉莉酸 和赤霉素的信号转导关键基因参与植物激素的响应。这些途径的靶基因,在不同的高 水平的研究论文里都得到了充分的实验(例如,通过启动子分析,转基因,凝胶阻滞 体外实验等)验证。
[0118]
本实施例采用实施例1提供的试剂盒和b-cut&tag方法对spl9进行分析鉴定:
[0119]
第一天:
[0120]
甲醛交联和细胞核提取(步骤1到11)
[0121]
1.将0.5克pspl9::3xflag-rspl9转基因拟南芥花序置于含有25毫升isob(f+)的50 毫升离心管中,抽真空固定10分钟。
[0122]
2.加入2.5毫升2m甘氨酸,温和混匀,继续抽真空5分钟,终止反应。
[0123]
3.用无菌水将固定后的花序材料清洗3次,然后用吸水纸除去多余的水。
[0124]
4.材料在液氮中碾磨成细腻的粉末。
[0125]
5.将碾磨后的花序置于50毫升离心管中,加入25毫升冰预冷的isob,轻柔混匀, 置于冰上轻晃5分钟,使材料均匀重悬,400x g离心力,4度离心5分钟,收集沉淀。
[0126]
6.去除上清,将材料重悬在10毫升isob(t+)中,轻柔混匀,将离心管置于冰上裂 解5-10分钟。
[0127]
7.用500目的细胞筛过滤裂解物,大的没有裂解的组织将留在细胞筛上,细胞核 则通过细胞筛孔隙,被收集在下方放置的培养皿中。
[0128]
8.将含有细胞核的裂解物收集在2毫升离心管中,400x g离心力,4度离心5分 钟,收集细胞核。
[0129]
9.去除上清,将所有细胞核重悬合并在1毫升wb(c+e+)中,400x g离心力,4 度离心5分钟,收集细胞核。至此,可以收集到~50μl体积的细胞核。
[0130]
10.去除上清,至此,可以收集到~50μl体积的细胞核。加入1毫升wb(c+e+)重悬细胞核,400
×
g离心力,4度离心5分钟,收集细胞核。
[0131]
11.重复第10步清洗步骤共计三次,最后一次清洗结束,去除上清后,继续400
×ꢀ
g离心力,4度离心2分钟,尽量吸除残余的上清。
[0132]
一抗孵育(步骤12到17)
[0133]
12.将50μl细胞核用1毫升ab(c+d+)重悬,置于冰上待用
[0134]
13.在1.5毫升离心管中设置如下反应:两个igg对照组,每个重复含有250μl 细胞核重悬液,2ug igg;两个抗体实验组,每个重复含有250μl细胞核重悬液,2uganti-flag抗体。轻柔混匀,水平翘板摇床,12rpm,4度,孵育过夜。
[0135]
14.将反应管从摇床取下,350x g离心力,4度离心4分钟,收集细胞核,移除 上清。
[0136]
15.加入800μl wb(c+d+)重悬细胞核,置于水平翘板摇床,室温12rpm孵洗5 分钟。
[0137]
16.将反应管从摇床取下,350x g离心力,4度离心4分钟,收集细胞核,移除 上清。
[0138]
17.350x g离心力,4度继续离心2分钟,尽量移除多余的上清。
[0139]
二抗孵育(步骤18到22)
[0140]
18.加入含有1ug二抗的250μl wb(c+d+)重悬细胞核,轻柔混匀,置于水平翘 板摇床,室温12rpm孵育1小时。
[0141]
19.将反应管从摇床取下,350x g离心力,4度离心4分钟,收集细胞核,移除 上清。
[0142]
20.加入800μl wb(c+d+)重悬细胞核,轻柔混匀,置于水平翘板摇床,室温12rpm 孵洗5分钟。
[0143]
21.将反应管从摇床取下,350x g离心力,4度离心4分钟,收集细胞核,移除 上清。
[0144]
22.重复步骤21到22孵洗步骤共三次,最后一次清洗完成后,350x g离心力,4 度继续离心2分钟,尽量移除多余的上清。转座酶孵育步骤23到27。
[0145]
23.加入含有8pmol转座酶的250μl tib,重悬细胞核,轻柔混匀,置于水平翘 板摇床,室温12rpm孵育3-4小时。
[0146]
24.将反应管从摇床取下,350x g离心力,4度离心4分钟,收集细胞核,移除 上清。
[0147]
25.加入800μl wb(c+d+)重悬细胞核,轻柔混匀,置于水平翘板摇床,室温 12rpm孵洗5分钟。
[0148]
26.将反应管从摇床取下,350x g离心力,4度离心4分钟,收集细胞核,移除 上清。
[0149]
27.重复步骤25到26孵洗步骤共三次,最后一次清洗完成后,350x g离心力,4 度继续离心2分钟,尽量移除多余的上清。
[0150]
片段化反应和解交联(步骤28到30)
[0151]
28.加入300μl tb重悬细胞核,37度水浴锅孵育1小时,进行片段化反应。
[0152]
29.每管加入15μl edta,15μl 20%sds,终止反应。
[0153]
30.反应管置于65度水浴锅过夜,进行解交联。
[0154]
dna提取(步骤31到38)
[0155]
31.加入300μl ctab植物dna提取液,65度水浴锅孵育39分钟,期间每隔 10分钟轻柔颠倒混匀。
[0156]
32.加入600μl酚:氯仿:异戊醇(25:24;1),充分颠倒混匀
[0157]
33.将预装有phase gel lock的离心管13000xg预离心2分钟,然后将样本转移到 phase gel lock离心管中,13000x g,4度离心10分钟。
[0158]
34.将上清(约600μl)转移到新的1.5毫升离心管中,加入600μl氯仿,充分颠 倒混匀13000x g,4度离心10分钟。
[0159]
35.将上清(约600μl)转移到新的1.5毫升离心管中,加入600μl异丙醇,2μl 共沉淀剂,充分混匀,置于-20度沉淀1小时。
[0160]
36.13000x g,4度离心10分钟。
[0161]
37.移除上清,用75%酒精洗涤dna沉淀,13000x g,4度离心5分钟。
[0162]
38.移除上清,随后13000x g,4度离继续瞬离30秒,尽量移除上清。抽真空干燥 2分钟,将dna重新溶解在150μl无菌水中。
[0163]
生物素链霉亲和素纯化(步骤39到46)
[0164]
39.将5-10μl链霉亲和素磁珠重悬在500μl 1
×
sa wb中,混匀,置于磁力架上 约5分钟收集磁珠。
[0165]
40.去除上清,将离心管从磁力架上取下,将磁珠重悬在500μl 1
×
sa wb混匀, 置于磁力架上约5分钟收集磁珠。去除上清。(第二次洗涤)。
[0166]
41.将离心管从磁力架上取下,将磁珠重悬在500μl 2
×
sa wb混匀,置于磁力 架上约5分钟收集磁珠。去除上清。
[0167]
42.将离心管从磁力架上取下,将磁珠重悬在150μl 2
×
sa wb加入步骤38得到 的150μldna样本,混匀,置于杂交炉中常温旋转孵育20-30分钟。
[0168]
43.孵育结束后,将离心管置于磁力架上约5分钟收集磁珠。去除上清。
[0169]
44.将离心管从磁力架上取下,将磁珠重悬在500μl 1
×
sa wb混匀,置于杂交 炉中常温旋转洗涤5分钟。随后将离心管置于磁力架上约5分钟收集磁珠。去除上清。
[0170]
45.重复步骤44清洗步骤共计3次。
[0171]
46.将磁珠重悬在30μl无菌水中,此时的产物为生物素化的dna片段与链霉亲和 素磁珠结合的混合物。
[0172]
二代测序建库(步骤47到50)
[0173]
47.在pcr管中设置如下反应:30μl来自步骤46的dna产物,12μl无菌水, 50μl 2
×
pcr mix,4μl n50x,4μl n70x。混合均匀,在上层滴入50μl石蜡油防治 蒸发。
[0174]
48.在pcr仪中设置如下程序:72度3分钟,98度30s,然后98度30s,60度30s, 72度30s,16-18个循环,最后72度延伸5分钟。
[0175]
49.pcr结束后将产物暂时置于冰上,取出3μl产物,凝胶电泳检测产物的浓度 和大小分布。如果没有检测到明显的条带富集,则酌情每轮增加2个循环。
[0176]
50.pcr扩增产物用1.2倍体积的dna纯化磁珠(例如,市购的ampure xp磁珠) 纯化回收。
[0177]
在该流程的研发过程中,由于生物素亲和素结合之后很难再分开,我们尝试采用 了300mm naoh在65℃处理10分钟对dna进行变性的方法回收单链dna,发现 后续还要进行ph调节,dna重新沉淀纯化,最后得率也不高。
[0178]
因此,我们尝试了磁珠原位pcr的方法,以结合了生物素化dna片段的链霉亲 和素磁珠为模版进行pcr反应,同时在pcr管上层加入石蜡油,并在pcr过程中每 2-3轮循环后轻弹混匀防止磁珠沉降,同时,严格控制磁珠用量,防止过量磁珠对pcr 造成抑制。
[0179]
根据上述建立的流程,我们对spl9过表达植物进行b-cut&tag实验。图4展示 了spl9 b-cut&tag-seq实验的过程中,实施生物素亲和素纯化步骤后,未存在与磁 珠结合的染色质(未被转座酶识别切割并生物素化的染色质)情况。这些未剪切的染 色质本质上不是spl9的靶位点,经过纯化步骤之后就被去除,而纯化的生物素标记 片段经过磁珠原位pcr后进行了高通量测序。
[0180]
经过生物信息学分析,结果得到了6383个富集的峰,涉及4476个靶基因(有的 靶基因富集出现不止一个富集的峰,因而结果里出现了峰的个数多于靶基因个数的情 况)。这些富集的峰的信号主要分布在基因转录起始位点的上游(启动子区域)(图5)。 非常重要的是,与chip结果类似,b-cut&tag准确地鉴定到了所有上述spl9参与 的相关发育及代谢途径的已经明确报道过的靶基因,包括非编码rna(mir172)、多个 mads box、myb、della和jaz基因,以及次生代谢产物花色素苷(f3’h)和蜡质 合成的关键基因(cer1)(图6)。
[0181]
此外,我们还鉴定到了一系列未被报道过的靶基因,在b-cut&tag和传统chip 结果中同时存在(共有的靶基因),这些通过不同的实验原理的技术都能同时稳定鉴定 到的靶基因,将是后续研究重点关注的研究对象,对研究spl9参与调控的新途径、 发挥的新功能具有非常好的提示作用。
[0182]
因此,通过本实施例的流程和以拟南芥spl9转录因子为例进行的实验,我们得 出
结论,b-cut&tag与二代测序结合(b-cut&tag-seq)可以很好地应用于植物转录 因子与dna互作的研究,特别适合从基因组范围内大规模筛选转录因子的靶基因位 点。
[0183]
实施例4应用b-cut&tag-qpcr进行特定基因位点组蛋白h3k3me3修饰水平的 研究
[0184]
本实施例对b-cut&tag后续是否可以与荧光定量pcr结合(b-cut&tag-qpcr), 发挥与传统的chip-qpcr同等的功能,用于鉴定转录因子与特定靶位点的结合,进行 了试验。
[0185]
以海岛棉叶片组蛋白h3k3me3修饰水平的为研究目标,进行b-cut&tag-qpcr 体系的探索。
[0186]
海岛棉叶片组蛋白h3k3me3修饰的b-cut&tag操作步骤与实施例3的操作步骤 类似,但在细胞核使用量上有所区别,组蛋白h3k3me3修饰细胞核使用量为100μl, 相应地,一些操作步骤中的缓冲液用量,抗体和转座酶的用量有所不同。并且,不需 要使用二抗进行信号放大。
[0187]
具体步骤为:
[0188]
进行实施例3中的步骤1到46步的操作,除了以下几个步骤有差别:
[0189]
步骤13.在1.5毫升离心管中设置如下反应:两个igg对照组,每个重复含有100μl 细胞核重悬液,1ug igg;两个抗h3k4me3抗体实验组,每个重复含有100μl细胞核 重悬液,1ug anti-h3k4me3抗体。轻柔混匀,水平翘板摇床,12rpm,4度,孵育过 夜。
[0190]
省略二抗孵育步骤18-20。
[0191]
步骤23.加入含有4pmol转座酶的100μl tib,重悬细胞核,轻柔混匀,置于水 平翘板摇床,室温12rpm孵育3-4小时。
[0192]
步骤38.移除上清,随后13000x g,4度离继续瞬离30秒,尽量移除上清。抽真 空干燥2分钟,将dna重新溶解在120μl无菌水中。取出20μl dna,记做“input”, 剩下的100μl加入50μl(1:0.5)dna纯化磁珠(如ampure xp beads磁珠),室温 结合20分钟,用磁力架分离磁珠,收集上清未被磁珠结合的小片段(~150μl)。
[0193]
随后将上述产物按照步骤到39到步骤45所述进行生物素亲和素纯化。
[0194]
步骤46中,在所得链霉亲和素磁珠-dna混合物中加入50μl eb缓冲液,90度 10分钟洗脱dna。用磁力架分离收集上清,产物用于后续qpcr分析。
[0195]
qpcr和数据分析(步骤51到55)
[0196]
51.接上述步骤46之后,根据chip-qpcr相似的引物设计原则设计靶标位点序列 特异的qpcr引物对,记作gspf和gspr。
[0197]
52.按照以下原则设置qpcr反应体系:模板为抗体组和igg对照组的dna,每 管体系加入1μl上述步骤46得到的dna;同时为抗体组和igg对照组的input dna 设置反应,模板为1μl步骤38中的input dna,记做1%input。为每个反应设置3 个技术重复。
[0198]
53.qpcr反应结束后,计算样本的扩增循环数(ct值)
[0199]
54.采用δct法计算靶序列在不同样本中的含量,igg对照组的ct均作为归一化 的对照。将抗体实验组的ct减去igg对照组的ct,δct=ct
抗体实验组-ct
igg
,则靶序列在 抗体实验组与igg对照组相比的富集倍数为2-δct
。
[0200]
55.或者,将样本自身的1%input的ct作为归一化的对照。δct=ct
样本
–
ct
1%input
; 样本指抗体组和igg对照组;则靶序列在样本中的含量占其1%input的比例为2-δct x 100%,比较抗体组与igg对照组该比例的差异。
[0201]
根据转座酶二聚体的包埋和工作规律,我们总结了染色质靶位点受到转座酶切割 发后,转座酶二聚体中两个的接头引物两两组合成后以四种不同的“黏贴”方式被加入 到片段化dna序列中(图7a,左)。
[0202]
为此,我们设计了公共接头引物p1(序列为attactaggtctcgtgggctcgg; qpcr,overlap with primer c),以及按照chip-qpcr引物设计原则设计靶标位点序列 特异的qpcr引物对,记作记作gspf和gspr,理论上,通过引物p1/gspr的组合, 可以鉴定场景1产生的目的片段;通过引物p1/gspf的组合,可以鉴定场景2产生的 目的片段;通过靶标位点特异引物对gspf和gspr的组合,可以鉴定场景1,场景2 和场景4的目的片段总和。场景3中,由于掺入的均为未生物素化的接头c,该部分 产物在生物素亲和素纯化的步骤丢失,不在qpcr能鉴定的范围之列(图7a,右)。
[0203]
我们利用靶标位点序列特异的qpcr引物对(gspf/gspr)进行h3k4me3修饰 b-cut&tag反应后的qpcr。以基因gb_d10g1774为例,我们分别选择了h3k4me3 修饰水平高的基因5’端(区域1),以及h3k4me3修饰水平高的基因3’下游(区域2), 分别设计了两对基因特异的gspf/gspr引物,引物序列如seq id no.24-seq idno.27所示。
[0204]
qpcr结果表明,与igg对照组相比,h3k4me3实验组的靶标区域的含量在区域 1处是对照组的10倍,而在区域2处,与对照组相比没有明显富集,这与 b-cut&tag-seq得到的片段读数(reads count)统计结果趋势一致(图7b)。同时,我 们也评估了另外一种归一化方法,即采用样本自身链霉素亲和纯化前的1%的inputdna作为归一化的对照,结果发现,h3k4me3实验组在区域1处靶标片段的比例为 其1%input对照的36.6%,而igg对照组在区域1处靶标片段的比例为其1%input对 照的3.59%;h3k4me3实验组在区域1的靶标片段发生了显著的富集。而在区域2处, h3k4me3实验组靶标片段的比例为其1%input对照的2.45%,而igg对照组靶标片段 的比例为其1%input对照的2.34%(图7c),h3k4me3实验组在区域2的靶标片段没有 明显的富集。
[0205]
由于基因特异的引物无法排除转座酶背景切割产生的生物素标记的大片段(》1000 bp),我们尝试了公用引物序列p1分别与靶标位点序列特异的gspf和gspr配对进行 h3k4me3修饰b-cut&tag反应后的qpcr的可行性。我们发现,分别来自切割场景 1和场景2的两条重叠或者部分重叠的dna片段,在pcr过程中仅p1引物存在条件 下发生非特异的pcr扩增(图8)。
[0206]
由此,不推荐使用公共引物p1与特异引物的组合进行qpcr。
[0207]
为了解决基因特异的引物无法排除转座酶背景切割产生的生物素标记的大片段 (》1000bp)的问题,我们在生物素亲和素纯化之前,增加了片段分选的步骤,将背 景切割产生的大片段通过dna纯化磁珠去除(步骤38)。
[0208]
因此,我们成功建立了b-cut&tag-qpcr的流程。
[0209]
实施例5应用b-cut&tag-qpcr进行转录因子与预测靶基因位点结合的研究
[0210]
最后,为了确定我们基于上述组蛋白修饰水平建立的b-cut&tag-qpcr流程是否 适用于验证转录因子的特异性结合,我们以拟南芥spl9和水稻磷元素代谢相关的重 要转录因子phr2为例,按照实施例1的转录因子的b-cut&tag操作流程及实施例4 中的qpcr流程,进行了植物转录因子的b-cut&tag-qpcr。由于我们使用的atspl9 和osphr2过表达的拟南芥和水稻均为3xflag标签融合蛋白,因此cut&tag体系 中仍然使用anti-flag抗体。
[0211]
首先应用b-cut&tag-qpcr确定拟南芥spl9的特异性结合。我们以三个文献中 已经报道并详细研究过的atspl9的靶基因,包括拟南芥毛状体发育中的attcl1、 attry(yu et al.,2010)和开花调控中的atful基因(wang et al.,2009),用于b-cut&tag-qpcr的基因特异性引物对与之前文献报道的chip-qpcr检测引物相同 (wang et al.,2009;yu et al.,2010),序列如seq id no.28-seq id no.35。以act2 基因作为对照。根据b-cut&tag-seq的数据,我们总结了定位到qpcr目标区域的片 段读数(图9a和b),attcl1、attry、atful和atact2的qpcr目标区域的rpm(每 百万reads中的比对到该区域的reads数)比值分别为2.45、2.25、3.18和1.45,一致地, b-cut&tag-qpcr结果显示,当rspl9检测组与igg对照组相比时,attcl1、attry、 atful和atact2目标区域富集了1.78、1.98、2.01和1.15倍(图9c),与高通量测序 的结果趋势一致。
[0212]
我们进一步以过表达osphr2的水稻植株为材料,应用b-cut&tag-qpcr确定了 osphr2的特异性结合。osphr2是一个重要的tf,在水稻中的磷酸盐稳态中起作用。 我们选择了osphr2的三个靶基因,包括低亲和性的pi转运体ospt2(liu et al.,2010), 它是osphr2的直接靶点,负责osphr2过表达介导的过量茎pi积累;高亲和力的pi 转运体ospt8,该基因也是pi稳态的关键(jia et al.,2011);am共生相关标记基因 osram1,该基因在菌根共生中受以osphr2为中心的网络的调控(shi et al.,2021)。 我们鉴定了phr1结合位点(p1bs)在ospt2、ospt8和osram1的启动子中的分布, 并设计qpcr引物其产物片段覆盖所指示的p1bs顺式元件(图9d),序列如seq idno.36-seq id no.41。在磷充足(+p)的培养条件下,osphr2实验组(抗flag标 签抗体)与igg对照组相比,ospt8和osram1的靶区均未富集,而ospt2的靶区富 集为igg对照组的2.36倍。相比之下,在磷缺失(-p)的条件下,ospt2、ospt8和osram1 的靶标区分别富集2.56倍、2.13倍和3.47倍(图9e),这些结果表明,在磷充足(+p) 和磷缺乏(-p)条件下,osphr2对磷平衡和菌根共生具有不同的调节作用。
[0213]
通过以上实施例,我们展示了本发明提供的cut&tag试剂盒及流程能够有效地 鉴定转录因子与dna的结合。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1