一种同步定量测定尿液中多类芳香族化合物与代谢产物浓度的方法
1.本发明属于毒害有机污染物分析技术领域,特别涉及一种同步定量测定尿液中多类芳香族化合物与代谢产物浓度的方法。
背景技术:
2.芳香族化合物,如硝基酚、氯酚等单环化合物,多环芳烃及其衍生物等多环化合物等,是环境中普遍存在的污染物,主要来自化石燃料的不完全燃烧,农药残留及工业废气废水废渣中。长期暴露于芳香族化合物可能会增加罹患疾病的风险,如暴露于硝基酚可引起神经系统及肝、肾等器官损伤;五氯酚的高暴露可能会增加自发性流产的风险;长期暴露于多环芳烃可引起肺癌等多种癌症发病率与死亡率倍增;研究表明:硝基多环芳烃、杂环芳烃等多环芳烃衍生物,在相对低浓度下,呈现出比原型多环芳烃相当或更高毒性。芳香族化合物可通过呼吸、饮食摄入和皮肤暴露等方式进入生物体内,在肝脏等器官中代谢成含羟基、羧基等官能团的产物,也可能以污染物原型形式存在于生物体内,并通过尿液等方式排出体外。因此,可通过同步检测尿液中的多类芳香族化合物与代谢产物的含量来评估生物体内芳香族化合物的暴露水平和负荷。
3.目前,各类芳香族化合物的分析检测技术已有很多成熟研究,如利用lc-ms/ms高通量技术检测尿液中硝基酚、氯酚、双酚类化合物、对羟基苯甲酸酯等多类单环芳香族化合物的分析方法;尿液中挥发性污染物暴露与效应标志物同步的检测技术报道,也有文献报道了一种血液中多环芳烃及其衍生物的同步检测技术。总结现有技术发现有以下几点不足:1)单环与多环芳香族化合物性质差异较大,缺乏一种全面同步分析检测这两类芳香族化合物的分析方法;2)有关多环芳烃衍生物的分析方法停留在血液等介质中污染物原型的分析方面。生物样品较难获取、且取样量有限,分类分析不同种类芳香族化合物,用样量较大、增加检测成本、耗时较长。因此,现有技术主要针对芳香族化合物暴露的全面评价还面临挑战,如何使用少许样品量同步分析多类芳香族化合物与代谢产物的分析技术迫在眉睫。
4.对于尿液中多类芳香族化合物与代谢产物的同步定量方法,其困难在于:1)芳香族化合物种类繁多,从单环至多环、苯环上取代基种类与数量不同等导致各物质间理化性质差异较大,选择一种合适的萃取溶剂同步萃取多类目标物是关键技术问题之一;2)液相色谱中物质种类较多,难以将各个化合物完全分离、单个化合物受物质总量影响,质谱灵敏度会有所下降。因此,如何平衡多类芳香族化合物与代谢产物在液相色谱串联三重四级杆质谱中的灵敏度也是一大难题。
技术实现要素:
5.为了克服现有技术中存在的缺点与不足,本发明的目的在于提供一种同步定量测定尿液中多类芳香族化合物与代谢产物浓度的方法。
6.本发明的目的通过下述技术方案实现:
7.一种同步定量测定尿液中多类芳香族化合物与代谢产物浓度的方法,包括以下操作步骤:
8.s1.取尿液样品加入同位素内标物后混匀,依次加入缓冲溶液和β-葡萄糖醛苷酶-芳基硫酸酯酶,恒温酶解;
9.s2.经过步骤s1酶解后的尿液用有机溶剂进行萃取,离心分离并收集上清液,再次加入有机溶剂重复萃取、离心操作,将所得上清液混合并浓缩定容至极性溶液中,得到样品浓缩液;
10.s3.采用液相色谱三重四级杆质谱对步骤s2所得样品浓缩液中的芳香族化合物与代谢产物进行分析,通过定量与数据处理,得到尿液中目标物多类芳香族化合物与代谢产物的浓度。
11.优选地,步骤s1中所述的同位素内标物为
13c6-4-氯邻苯二酚、
13c6-2,4,5,6-四氯酚、2-羟基萘-d7、1-氨基萘-d7、2-羟基芴-d9、
13c6-3-羟基菲、1-羟基芘-d9、1-氨基芘-d9和3-羟基苯并[a]芘-d
11
中的一种以上;所述的同位素内标物质量为1~10ng。
[0012]
优选地,步骤s1中所述的缓冲溶液为磷酸氢二钠-氢氧化钠溶液、磷酸-乙酸铵溶液、乙酸钠-乙酸溶液、乙酸铵-乙酸溶液或磷酸二氢钠-磷酸溶液;所述的缓冲溶液的浓度为0.1~10mol/l,ph值为1~14。
[0013]
优选地,步骤s1中所述的尿液样品、缓冲溶液及β-葡萄糖醛苷酶-芳基硫酸酯酶的体积比为(50~1000):(20~500):1;所述的酶解温度为20~50℃,酶解时间为0.5~24h。
[0014]
优选地,步骤s2中所述的有机溶剂为乙酸乙酯、甲基叔丁基醚、二氯甲烷、正己烷、环己烷和异辛烷中的一种以上;所述有机溶剂与步骤s1中所述的尿液样品的体积比为(0.5~6):1。
[0015]
优选地,步骤s2中所述的离心的转速为1000~8000rpm,离心的时间为3~30min;所述重复的次数为1~5次;所述极性溶液为超纯水、甲醇和乙腈中的一种以上。
[0016]
优选地,步骤s3中所述的液相色谱三重四级杆的测试条件为:所用液相色谱流动相水相为甲酸水溶液、乙酸水溶液或乙酸铵水溶液,其浓度为0.005~50g/l;有机相为甲醇或乙腈;柱温为20~50℃,梯度洗脱程序开始为1~5%有机相,20min内升至99%有机相,再10min内降至5%有机相,保留0~10min;使用c18反相色谱柱分离目标物,离子源为电喷雾电离源或大气压化学电离源,离子源中氮气温度为200~400℃,流速为1~10l/min,鞘气温度为200~500℃,流速为5~20l/min,毛细管电压为2000~5000v。
[0017]
优选地,步骤s3中所述的目标物为4种硝基酚、8种氯酚、12种羟基多环芳烃、2种羧基多环芳烃、5种羟基杂环芳烃、3种羟基硝基多环芳烃和5种氨基多环芳烃。
[0018]
优选地,所述4种硝基酚分别为邻硝基酚、间硝基酚、对硝基酚和3-甲基-4-硝基酚;所述8种氯酚分别为4-氯邻苯二酚、2-一氯酚、3-一氯酚、4-一氯酚、2,3,5,6-四氯酚、2,4,5,6-四氯酚、2,3,4,5-四氯酚和五氯酚;所述12种羟基多环芳烃分别为1-羟基萘、2-羟基萘、2-羟基芴、3-羟基芴、1-羟基菲、2-羟基菲、3-羟基菲、4-羟基菲、9-羟基菲、1-羟基芘、6-羟基和3-羟基苯并[a]芘;所述2种羧基多环芳烃分别为2-萘甲酸和1-芘甲酸;所述5种羟基杂环芳烃分别为5-羟基异喹啉、8-羟基喹啉、4-羟基咔唑、3-羟基咔唑和2-羟基氧芴;所述3种羟基硝基多环芳烃分别为4-硝基-1-羟基萘、6-硝基-1-羟基芘和3-硝基-1-羟基芘;
所述5种氨基多环芳烃分别为1-氨基萘、2-氨基萘、2-氨基芴、1-氨基芘和6-氨基。
[0019]
优选地,步骤s3中所述的定量是使用与液相色谱三重四级杆质谱配套的定量软件进行;所述的数据处理过程如下公式:
[0020]
c=cq×vc
/vs[0021]
式中:
[0022]
c是指样品中目标物的含量,单位为纳克每毫升(ng/ml);
[0023]cq
是指定量软件定量所得样品中目标物的含量,单位为纳克每毫升(ng/ml);
[0024]vc
是指样品浓缩液体积,单位为毫升(ml);
[0025]vs
是指最初使用的样品体积,单位为毫升(ml)。
[0026]
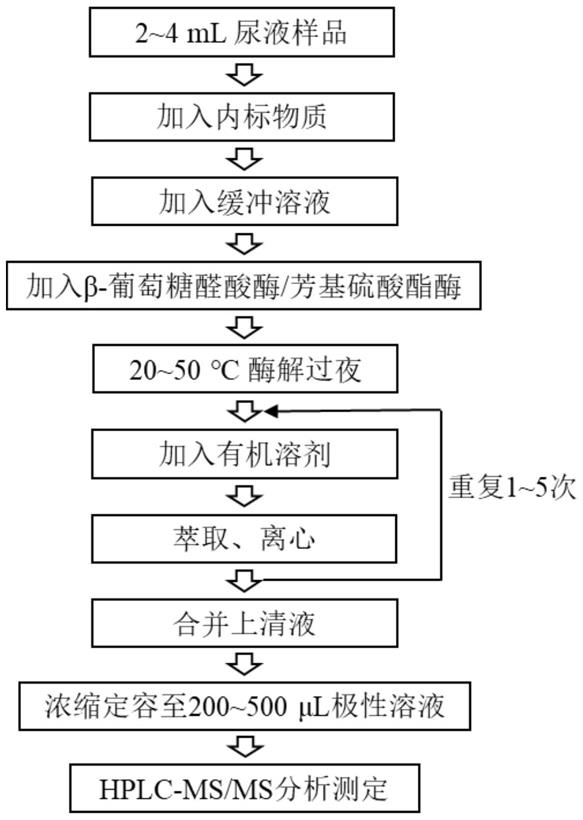
上述同步定量测定尿液中芳香族化合物与代谢产物浓度的方法的流程图如图1所示。
[0027]
本发明相对于现有技术具有如下的优点及效果:
[0028]
(1)本发明可同步定量测定1~5环芳香族化合物与代谢产物,物质范围广,可以进行多类芳香族化合物与代谢产物的一次分析。
[0029]
(2)本发明简化了样品前处理步骤,增加了可同步处理的样品量,缩短了样品分析所需时间。
[0030]
(3)本发明通过同位素内标法进行定量,减少了可能的基质干扰,提高了定量的准确性。
[0031]
(4)本发明操作简单,便于在不同实验室开展大量尿液样品的分析。
[0032]
(5)本发明方法兼容性强,在进行方法适用性验证后,留存的样品均可用于结构及理化性质相似的化合物及代谢产物(如对羟基苯甲酸酯、邻苯二甲酸单酯等)的分析。
[0033]
(6)本发明可实现对尿液中多类芳香族化合物与代谢产物的同步检测,为评估生物体内芳香族化合物的暴露水平和负荷提供有效的方法手段。
附图说明
[0034]
图1为本发明同步定量测定尿液中芳香族化合物与代谢产物浓度的方法的流程图。
[0035]
图2为流动相为甲酸与甲醇时4种目标物的出峰效果。
[0036]
图3为乙酸乙酯为萃取溶剂时16种目标物的回收率。
[0037]
图4为正己烷/二氯甲烷(3/1,v/v)混合液为萃取溶剂时9种目标物的回收率。
具体实施方式
[0038]
下面结合具体实施例及附图进一步说明本发明的内容,但不应理解为对本发明的限制。若未特别指明,实施例中所用的技术手段为本领域技术人员所熟知的常规手段。除非特别说明,本发明采用的试剂、方法和设备为本技术领域常规试剂、方法和设备。
[0039]
以下所有实施例测试均采用液相色谱三重四级杆质谱(lc-ms/ms)进行,选择甲酸水溶液、乙酸水溶液或乙酸铵水溶液等不同溶液为水相(具体见各实施例),以甲醇为有机相,流速为0.4ml/min;柱温为40℃;梯度洗脱程序开始5%甲醇,0-2min,5-15%甲醇;2-2.1min,15-50%甲醇;2.1-10min,50%甲醇;10-15min,50-80%甲醇;15-18min,80-95%甲
醇;18-19min,95%甲醇;19-20min,95-99%甲醇;20-22min,99%甲醇;22.1-30min,99-5%甲醇;后运行4min。使用c18反相色谱柱(4.6mm
×
100mm,2.7μm)分离目标物;离子源为喷射流电喷雾电离源(ajs esi)正负离子模式,离子源中氮气温度为300℃,流速为5l/min,鞘气温度为350℃,流速为12l/min,毛细管电压为3500v。
[0040]
实施例1
[0041]
将甲酸水溶液与甲醇作为流动相时的液相色谱三重四级杆质谱时的物质出峰效果——以1-氨基萘(1-anap)、2-氨基萘(2-anap)、1-羟基萘(1-oh-nap)与2-羟基萘(2-oh-nap)为例。
[0042]
本实施例lc-ms/ms流动相选择1.22g/l甲酸水溶液为水相,以甲醇为有机相。
[0043]
图2为浓度为50ng/ml的1-anap、2-anap、1-oh-nap与2-oh-nap在上述lc-ms/ms参数设置下的相应信号,可见,1-anap与2-anap在该参数设置下,相应信号非常强,能达到106级别,而1-oh-nap与2-oh-nap的信号则低至102,通常尿液中1/2-oh-nap的浓度为10-2
~103ng/ml级别,显然,此种参数设置条件下,虽然能满足1-anap与2-anap的分析测定条件,但无法同时满足1-oh-nap与2-oh-nap的分析测定需求。
[0044]
实施例2
[0045]
使用乙酸乙酯作为有机溶剂萃取尿液中16种目标物芳香族化合物——以2,3,5,6-四氯酚(2,3,5,6-tetrachlorophenol,2,3,5,6-tetracp)、2,4,5,6-四氯酚(2,4,5,6-tetrachlorophenol,2,4,5,6-tetracp)、2,3,4,5-四氯酚(2,3,4,5-tetrachlorophenol,2,3,4,5-tetracp)、五氯酚(pcp)、1-oh-nap、2-oh-nap、2-羟基芴(2-oh-flu)、3-羟基芴(3-oh-flu)、1-羟基菲(1-oh-phe)、2-羟基菲(2-oh-phe)、3-羟基菲(3-oh-phe)、4-羟基菲(4-oh-phe)、9-羟基菲(9-oh-phe)、1-羟基芘(1-oh-pyr)、3-羟基咔唑(3-oh-cbz)及2-羟基氧芴(2-oh-dbf)为例。
[0046]
本实施例采用基质尿液样品进行回收率实验。即随机采集30例尿液样品,混合并稀释10倍,得到基质尿液样品,保存至-80℃冰箱中。本实施例采用1个基质空白样品和3个平行基质加标样品进行回收率实验。
[0047]
基质空白样品,是指利用基质尿液样品,按照实际尿液样品的步骤进行实验所得的样品;基质加标样品,是指利用基质尿液样品,加入已知质量的目标物,再按照实际尿液样品的步骤进行实验所得的样品。
[0048]
本实施例样品前处理方法如下:
[0049]
将基质尿液样品解冻,取2ml尿液,加入20ng上述16种目标物(仅添加于基质加标样品中,不加至基质空白样品中),依次加入2~4ng同位素内标物(
13c6-2,4,5,6-tetracp、2-oh-nap-d7、2-oh-flu-d9、
13c6-3-oh-phe及1-oh-pyr-d9)、1ml 1mol/l的乙酸钠-乙酸水溶液(ph=4~6)及5μlβ-葡萄糖醛苷酶-芳基硫酸酯酶,混匀,在37℃恒温摇床上酶解2~12h。
[0050]
酶解后的样品中加入4ml乙酸乙酯进行萃取,离心(2500rpm,5min)移取上清液,重复以上操作3次,共收集12ml上清液,浓缩并定容至200μl甲醇中,得到样品浓缩液,采用液相色谱三重四级杆质谱对目标物进行分析。通过定量软件进行定量,经如下公式进行数据处理,得到尿液中目标物的回收率。
[0051]
rec=c
spk_1
/c
spk_2
×
100%=[(c
q_spk_1
–cq_blk
)
×vc
/vs]/(c
spk_2
×vc
/vs)
×
100%=(c
q_spk_1
–cq_blk
)/c
spk_2
×
100%
[0052]
式中:
[0053]
rec是指基质加标样品的回收率(%);
[0054]cspk_1
是指基质加标样品中实验所得的目标物含量,单位为纳克每毫升(ng/ml);
[0055]cspk_2
是指基质加标样品中实际加入的目标物含量,单位为纳克每毫升(ng/ml);
[0056]cq_spk_1
是指定量软件定量所得基质加标样品中目标物的含量,单位为纳克每毫升(ng/ml);
[0057]cq_blk
是指定量软件定量所得基质空白样品中目标物的含量,单位为纳克每毫升(ng/ml);
[0058]vc
是指样品浓缩液体积,0.2ml;
[0059]vs
是指最初使用的样品体积,2ml。
[0060]
本实施例lc-ms/ms流动相选择0.39g/l乙酸铵水溶液为水相,以甲醇为有机相。
[0061]
图3为在上述条件下的16种芳香族化合物的回收率。可以看出,在用乙酸乙酯作为萃取溶剂时,对于4种氯酚的效果并不理想,回收率仅为52.0
±
2.10%至56.5
±
1.57%之间;其他12种芳香族化合物的回收率在73.5
±
1.71%与99.4
±
6.34%之间,满足美国环保署推荐的70~130%之间。此实施例说明,乙酸乙酯不适用于萃取高氯苯酚类物质。
[0062]
实施例3
[0063]
使用正己烷/二氯甲烷(3/1,v/v)混合液作为有机溶剂萃取尿液中芳香族化合物——以邻硝基酚(2-np)、间硝基酚(3-np)、对硝基酚(4-np)、3-甲基-4-硝基酚(3-m-4-np)、4-氯邻苯二酚(4-cct)、2,3,5,6-tetracp、2,4,5,6-tetracp、2,3,4,5-tetracp及pcp为例。
[0064]
本实施例采用基质尿液样品(制备方法同实施例2)进行回收率实验。本实施例采用1个基质空白样品和3个平行基质加标样品进行回收率实验。基质空白样品与基质加标样品定义见实施例2。
[0065]
本实施例样品前处理方法如下:
[0066]
将基质尿液样品解冻,取2ml尿液,加入20ng上述9种目标物(仅添加于基质加标样品中,不加至基质空白样品中),依次加入2ng同位素内标物(
13c6-4-cct与
13c6-2,4,5,6-tetracp)、1ml 1mol/l的乙酸铵-乙酸水溶液(ph=4~6)及5μlβ-葡萄糖醛苷酶-芳基硫酸酯酶,混匀,在37℃恒温摇床上酶解2~12h。
[0067]
酶解后的样品中加入4ml正己烷/二氯甲烷(3/1,v/v)混合液进行萃取,离心(3000rpm,5min)移取上清液,重复以上操作3次,共收集12ml上清液,浓缩并定容至200μl甲醇中,得到样品浓缩液,采用液相色谱三重四级杆质谱对目标物进行分析。通过定量软件定量及数据处理(公式同实施例2),得到尿液中目标物的回收率。
[0068]
本实施例lc-ms/ms流动相选择1.05g/l乙酸水溶液为水相,以甲醇为有机相。
[0069]
图4为在上述条件下的9种芳香族化合物的回收率。可以看出,在用正己烷/二氯甲烷(3/1,v/v)混合液作为萃取溶剂时,对于4种高氯酚的萃取效果显著优于实施例2,回收率在91.0
±
2.07%与92.1
±
2.80%之间;3-m-4-np的回收率也较好,为74.4
±
0.47%;但2/4-np、3-np及4-cct的回收率很低,分别为43.1
±
2.92%、51.1
±
0.57%与14.6
±
1.25%。远达不到70~130%的要求。此实施例说明,正己烷/二氯甲烷(3/1,v/v)混合液不适用于萃取2-np、3-np、4-np及4-cct。
[0070]
实施例4
[0071]
按照实施例2的萃取溶剂与仪器分析条件,对本发明进行实际应用——以1-oh-nap、2-oh-nap、2-oh-flu、3-oh-flu、1-oh-phe、2-oh-phe、3-oh-phe、4-oh-phe、9-oh-phe、1-oh-pyr、3-oh-cbz及2-oh-dbf为例。
[0072]
一种同步定量测定尿液中芳香族化合物与代谢产物浓度的方法及其应用,采用以下条件进行测定:
[0073]
本实施例尿液样品采自我国某焦化厂在职工人与远离焦化污染的对照区居民,各10例样品。样品采集完成后转移至实验室,密封保存于-80℃冰箱中。
[0074]
本实施例样品前处理步骤如下:
[0075]
将尿液样品解冻,取2ml尿液,依次加入2~4ng同位素内标物(2-oh-nap-d7、2-oh-flu-d9、
13c6-3-oh-phe及1-oh-pyr-d9)、1ml 1mol/l的乙酸钠-乙酸水溶液(ph=4~6)及5μlβ-葡萄糖醛苷酶-芳基硫酸酯酶,混匀,在37℃恒温摇床上酶解2~12h。
[0076]
酶解后的样品中加入4ml乙酸乙酯进行萃取,离心(2500rpm,5min)移取上清液,重复以上操作3次,共收集12ml上清液,浓缩并定容至200μl甲醇中,得到样品浓缩液,采用液相色谱三重四级杆质谱对目标物进行分析。通过定量软件定量及如下公式进行数据处理,得到尿液中目标物的含量水平。
[0077]
c=cq×vc
/vs[0078]
c是指样品中目标物的含量,单位为纳克每毫升(ng/ml);
[0079]cq
是指定量软件定量所得样品中目标物的含量,单位为纳克每毫升(ng/ml);
[0080]vc
是指样品浓缩液体积,0.2ml;
[0081]vs
是指最初使用的样品体积,2ml。
[0082]
经样品前处理及仪器分析测定,我们对焦化工人与对照区居民尿液样品进行了分析,发现1-oh-nap、2-oh-nap、2-oh-flu、3-oh-flu、1-oh-phe、2-oh-phe、3-oh-phe、4-oh-phe、9-oh-phe、1-oh-pyr、3-oh-cbz及2-oh-dbf等12种目标物的总浓度显著高于对照区居民,分别为554
±
242ng/ml与199
±
92.7ng/ml。焦化工人尿液中各物质浓度为对照区居民的2.2~7.2倍。以上表明,焦化污染是pahs及其衍生物的重要来源。
[0083]
上述4个实施例为本发明以不同芳香族化合物与代谢产物为例在不同实验条件设定下呈现的不同效果的实施方式,本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所做的改变、修饰、替代、组合和简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1