一种3-氨基-6-甲氧基哒嗪有关物质的检测方法与流程
1.本发明属于医药技术领域,涉及一种医药中间体3-氨基-6-甲氧基哒嗪中有关物质的检测方法。
背景技术:
2.3-氨基-6-甲氧基哒嗪(英文名:3-amino-6-methoxydazine)是一种重要医药中间体,其可以用于具有显著生物活性的吡啶并嘧啶类化合物或药品,如抗癌药瑞卢戈利,该药可用于治疗子宫内膜异位症引起的疼痛、子宫肌瘤以及晚期前列腺癌等适应症。
3.目前,关于该化合物制备方法的报道有很多,在现有合成工艺的过程中往往会引入多种工艺杂质,如3-氨基-6-氯哒嗪(杂质a)、3-氨基-6-羟基哒嗪(杂质b)、3-氯-6-羟基哒嗪(杂质c)、3,3-二氯哒嗪(杂质d)、3,6-二甲氧基哒嗪(杂质e)、3-氨基-6-甲氧基哒嗪(杂质f)、3-氨基哒嗪(杂质g)等7种,但并没有相关文献报道这些工艺杂质的检测方法。因此,开发一种能够实现对3-氨基-6-甲氧基哒嗪中有关物质的检测方法,对提供用药安全和3-氨基-6-甲氧基哒嗪的质量控制具有重要意义。
技术实现要素:
4.发明目的:本发明旨在提供一种分离度、准确度、重现性及稳定性均良好,检出限低,分析时间短的3-氨基-6-甲氧基哒嗪中有关物质的检测方法。
5.技术方案:本发明提供的3-氨基-6-甲氧基哒嗪中有关物质的检测方法包含以下步骤:
6.(1)配制供试品溶液和对照溶液;
7.(2)检测和记录:采用液相色谱法对所述供试品溶液和对照溶液进行检测并记录谱图,所述液相色谱的色谱条件为:
8.色谱柱:耐水的十八烷基键合硅胶柱;流动相:以六氟磷酸钾缓冲液(用磷酸调ph至2.8~3.2)为流动相a,以乙腈为流动相b,梯度洗脱;流速:0.9~1.1ml/min;柱温:33~37℃;检测波长:213~217nm。
9.作为优选,在液相色谱仪的进样器前接有鬼峰捕集柱。
10.进一步地,步骤(1)中供试品溶液的浓度为0.5mg/ml,对照溶液的浓度为1μg/ml。
11.溶剂:水。
12.溶液的配制方法:
13.杂质a贮备液(7.5μg/ml):精密称取15mg的杂质a对照品,置100ml量瓶中,加20ml 20%乙腈超声使溶解,冷却至室温,用水稀释至刻度,摇匀;精密量取该溶液5ml,置100ml量瓶中,用水稀释至刻度,摇匀。
14.其它杂质贮备液(7.5μg/ml):精密称取15mg的其它各杂质对照品,精密称定,置100ml量瓶中,加水超声使溶解,冷却至室温,用水稀释至刻度,摇匀;精密量取该溶液5ml,置100ml量瓶中,用水稀释至刻度,摇匀。
15.供试品溶液:精密称取3-氨基-6-甲氧基哒嗪供试品约25mg,置于50ml量瓶中,加水超声使溶解,再加水至刻度,摇匀;
16.对照溶液:量取上述供试品溶液2ml,置100ml量瓶中,用水稀释至刻度,摇匀,得溶液i;量取溶液i 5ml,置50ml量瓶中,用水稀释至刻度,摇匀。
17.进一步地,步骤(2)中检测使用的色谱柱的型号为atlantis t3柱或sepax hp c18柱。atlantis t3柱的规格为粒径(μm)
×
内径(mm)
×
柱长(mm):3
×
4.6
×
150;sepax hp c18柱的规格为粒径(μm)
×
内径(mm)
×
柱长(mm):5
×
4.6
×
250。
18.进一步地,步骤(2)流动相a中六氟磷酸钾缓冲液的浓度为15~25mmol/l。
19.进一步地,步骤(2)流动相梯度洗脱过程如下:
20.时间(min)流动相a(%)流动相b(%)010005100025752525.011000351000
21.进一步地,步骤(2)中检测的对照溶液、供试品溶液进样量为5~10μl。
22.计算公式:
[0023][0024][0025]
总杂(%)=未知单杂之和(%)+已知杂质之和(%);
[0026]
其中a
杂质
—供试品溶液中各已知杂质的峰面积;a
对照
—对照溶液峰面积;f—各已知杂质校正因子。
[0027]
有益效果:与现有技术相比,本发明具有如下显著优点:
[0028]
(1)建立了3-氨基-6-甲氧基哒嗪中有关物质的检测方法,可以快速、准确地测定各有关物质的含量;
[0029]
(2)方法操作简便,可以有效检测3-氨基-6-甲氧基哒嗪中有关物质的含量,灵敏度高(检出限为0.045μg/ml,定量限为0.15μg/ml)、专属性强(空白溶剂和降解实验对杂质检测干扰)、线性相关性良好(r2>0.999)、准确度高(不同浓度水平加标回收率在94%~106%之间,且回收率rsd值<5%,重复性及中间精密度rsd值均<3%);
[0030]
(3)方法耐用性好,可用于各种供试品中3-氨基-6-甲氧基哒嗪中有关物质的质量控制。
附图说明
[0031]
图1为系统适用性溶液的hplc色谱图。
[0032]
图2为空白溶剂的hplc色谱图。
[0033]
图3为供试品溶液的hplc色谱图。
[0034]
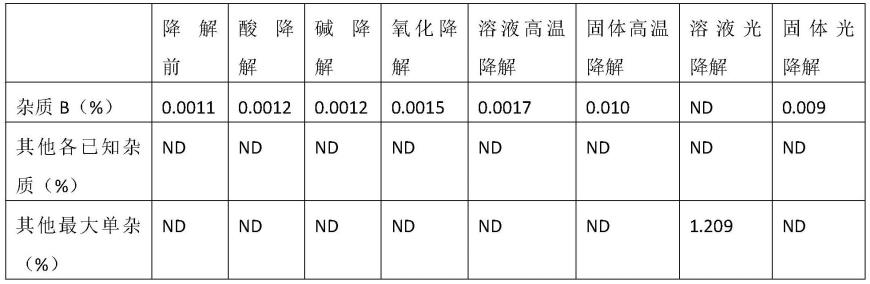
图4为对比实施例1中系统适用性溶液的hplc色谱图。
[0035]
图5为对比实施例2中系统适用性溶液的hplc色谱图
[0036]
图6为ph=2.7的色谱条件下系统适用性溶液的hplc色谱图。
[0037]
图7为ph=3.3的色谱条件下系统适用性溶液的hplc色谱图。
具体实施方式
[0038]
下面结合实施例和附图对本发明的技术方案作进一步说明。
[0039]
溶液的配制方法:
[0040]
杂质a贮备液(7.5μg/ml):精密称取15mg的杂质a对照品,置100ml量瓶中,加20ml 20%乙腈超声使溶解,冷却至室温,用水稀释至刻度,摇匀;精密量取该溶液5ml,置100ml量瓶中,用水稀释至刻度,摇匀。
[0041]
其它杂质贮备液(7.5μg/ml):精密称取15mg的其它各杂质对照品,精密称定,置100ml量瓶中,加水超声使溶解,冷却至室温,用水稀释至刻度,摇匀;精密量取该溶液5ml,置100ml量瓶中,用水稀释至刻度,摇匀。
[0042]
3-氨基-6-甲氧基哒嗪对照品贮备液的配制:精密称取3-氨基-6甲氧基哒嗪对照品约25mg,置50ml量瓶中,加水超声使溶解,冷却至室温,再用水稀释至刻度,摇匀。
[0043]
供试品溶液的配制:精密称取3-氨基-6-甲氧基哒嗪供试品约25mg,置于50ml量瓶中,加水超声使溶解,再加水至刻度,摇匀。
[0044]
对照溶液的配制:量取上述供试品溶液2ml,置100ml量瓶中,用水稀释至刻度,摇匀,得溶液i;量取溶液i 5ml,置50ml量瓶中,用水稀释至刻度,摇匀。
[0045]
实施例1系统适用性试验
[0046]
系统适用性溶液的配制:取3-氨基-6-甲氧基哒嗪供试品约25mg,精密称定,置50ml量瓶中,加水超声使溶解,冷却至室温,再精密加入7.5μg/ml的各杂质贮备液各5ml,用水稀释至刻度,摇匀,制成各杂质0.75μg/ml和3-氨基-6-甲氧基哒嗪0.5mg/ml的系统适用性溶液。进样测试,检测条件如下:色谱柱:atlantis t3柱;流动相:以20mmol/l六氟磷酸钾缓冲液(用磷酸调ph至3.0)为流动相a,以乙腈为流动相b,梯度洗脱;流速:1.0ml/min;柱温:35℃;检测波长:215nm;进样量5μl。记录结果,如表1和图1所示。
[0047]
表1系统适用性试验结果
[0048]
成分浓度(μg/ml)相对保留时间分离度杂质a(impuritya)0.750.8435.05杂质b(impurityb)0.750.25n/a杂质c(impurityc)0.750.957.34杂质d(impurityd)0.751.3719.88杂质e(impuritye)0.751.475.92杂质f(impurityf)0.751.533.47杂质g(impurityg)0.750.282.943-氨基-6-甲氧基哒嗪50012.64
[0049]
试验结果显示,系统适用性溶液中主峰与相邻杂质分离较好,各杂质间分离较好。
[0050]
实施例2专属性试验
[0051]
2.1干扰试验
[0052]
空白溶剂:水
[0053]
供试品溶液的配制:精密称取3-氨基-6-甲氧基哒嗪供试品约25mg,置50ml量瓶中,加水超声使溶液,冷却至室温,再用水稀释至刻度,摇匀。
[0054]
对空白溶剂和供试品溶液分别进样,检测条件如下:色谱柱:atlantis t3柱;流动相:以20mmol/l六氟磷酸钾缓冲液(用磷酸调ph至3.0)为流动相a,以乙腈为流动相b,梯度洗脱;流速:1.0ml/min;柱温:35℃;检测波长:215nm;进样量5μl。记录结果,谱图分别如图2和图3所示。结果表明,空白溶剂不干扰供试品的检测,且主峰纯度>990。
[0055]
2.2破坏试验
[0056]
供试品溶液:精密称取3-氨基-6-甲氧基哒嗪供试品约25mg,置于50ml量瓶中,加水超声使溶解,再加水至刻度,摇匀;
[0057]
对照溶液:量取上述供试品溶液2ml,置100ml量瓶中,用水稀释至刻度,摇匀,得溶液i;量取溶液i 5ml,置50ml量瓶中,用水稀释至刻度,摇匀。
[0058]
氧化降解试验:精密称取3-氨基-6-甲氧基哒供试品约25mg,置50ml量瓶中,加水超声使溶解,冷却至室温,加入3%过氧化氢溶液1ml,室温放置3小时,用水稀释至刻度,摇匀,即得。
[0059]
酸降解试验:取3-氨基-6-甲氧基哒供试品约25mg,精密称定,置50ml量瓶中,加水适量(约30ml)超声使溶解,冷却至室温,加0.1mol/l盐酸溶液1ml,室温放置3小时,用1ml的0.1mol/l氢氧化钠溶液中和,用水稀释至刻度,摇匀,即得。
[0060]
碱破坏:取3-氨基-6-甲氧基哒供试品约25mg,精密称定,置50ml量瓶中,加水适量(约30ml)超声使溶解,冷却至室温,加0.1mol/l氢氧化钠溶液1ml,室温放置3小时,用1ml的0.1mol/l盐酸溶液中和,用水稀释至刻度,摇匀,即得。
[0061]
固体高温破坏:取3-氨基-6-甲氧基哒供试品适量于称量瓶中,摊成≤5mm厚的薄层,置60℃的烘箱中放置3小时,取出,放冷至室温,取本品约25mg,精密称定,置50ml量瓶中,加水超声使溶解,冷却至室温,用水稀释至刻度,摇匀,即得。
[0062]
溶液高温破坏:取3-氨基-6-甲氧基哒供试品约25mg,精密称定,置50ml量瓶中,加水超声使溶解,冷却至室温,水浴60℃放置3小时,取出,冷却至室温,用水稀释至刻度,摇匀,即得。
[0063]
固体光破坏:取3-氨基-6-甲氧基哒供试品适量于称量瓶中,摊成≤5mm厚的薄层,置4500lx
±
500lx照度下放置3天,取出,取本品约25mg,精密称定,置50ml量瓶中,加水超声使溶解,冷却至室温,用水稀释至刻度,摇匀,即得。
[0064]
溶液光破坏:取3-氨基-6-甲氧基哒供试品约25mg,精密称定,置50ml量瓶中,加水超声使溶解,冷却至室温,置4500lx
±
500lx照度下放置1天,取出,用水稀释至刻度,摇匀,即得。
[0065]
将供试品溶液、对照溶液、降解后的样品分别进样,检测条件如下:色谱柱:atlantis t3柱;流动相:以20mmol/l六氟磷酸钾缓冲液(用磷酸调ph至3.0)为流动相a,以乙腈为流动相b,梯度洗脱;流速:1.0ml/min;柱温:35℃;检测波长:215nm;进样量5μl。记录结果,结果如下表2所示。
[0066]
表2强制降解试验结果
[0067][0068][0069]
计算公式:
[0070][0071][0072]
总杂(%)=未知单杂之和(%)+已知杂质之和(%);
[0073]
其中a
杂质
—供试品溶液中各已知杂质的峰面积;a
对照
—对照溶液峰面积;f—各已知杂质校正因子。
[0074]
结果表明,溶液光破坏条件下有降解杂质生成,但降解杂质并不干扰检测,分离度均符合要求,物料守恒,其他各条件3-氨基-6-甲氧基哒嗪均比较稳定。
[0075]
实施例3检出限和定量限
[0076]
3-氨基-6-甲氧基哒嗪对照品贮备液的配制:精密称取3-氨基-6甲氧基哒嗪对照品约25mg,置50ml量瓶中,加水超声使溶解,冷却至室温,再用水稀释至刻度,摇匀。
[0077]
定量限贮备液的配制:精密量取3-氨基-6-甲氧基哒嗪对照品贮备液1.5ml,其他各杂质贮备液5ml置同一100ml量瓶中,加水稀释至刻度,摇匀。
[0078]
定量限溶液的配制:精密量取定量限贮备液1ml,置50ml量瓶中,加水稀释至刻度,摇匀。
[0079]
检测限溶液的配制:精密量取定量限溶液3ml置10ml量瓶中,加水稀释至刻度,摇匀。
[0080]
将定量限溶液和检测限溶液分别进样,检测条件如下:色谱柱:atlantis t3柱;流动相:以20mmol/l六氟磷酸钾缓冲液(用磷酸调ph至3.0)为流动相a,以乙腈为流动相b,梯度洗脱;流速:1.0ml/min;柱温:35℃;检测波长:215nm;进样量5μl。记录结果,如表3所示。
[0081]
表3检出限与定量限结果汇总
[0082]
成分定量限(μg/ml)检出限(μg/ml)检出限s/n杂质a(impuritya)0.1550.04614.1杂质b(impurityb)0.1510.04526.2杂质c(impurityc)0.1550.04612.0杂质d(impurityd)0.1570.0475.7杂质e(impuritye)0.1550.0467.2杂质f(impurityf)0.1450.04411.6杂质g(impurityg)0.1520.0467.73-氨基-6-甲氧基哒嗪0.1500.04513.9
[0083]
从表中数据可知,各成分的定量限和检出限相当于供试品浓度的0.03%和0.009%。检出限和定量限都较低,方法的灵敏度高。
[0084]
实施例4线性范围和校正因子试验
[0085]
线性贮备液的配制:精密量取3-氨基-6-甲氧基哒嗪对照品贮备液1.5ml,其他各杂质贮备液5ml置同一100ml量瓶中,加水稀释至刻度,摇匀。
[0086]
线性-200%浓度溶液的配制:精密量取线性贮备液10ml,置50ml量瓶中,用水稀释至刻度,摇匀。
[0087]
线性-150%浓度溶液的配制:精密量取线性贮备液7.5ml,置50ml量瓶中,用水稀释至刻度,摇匀。
[0088]
线性-100%浓度溶液的配制:精密量取线性贮备液5ml,置50ml量瓶中,用水稀释至刻度,摇匀。
[0089]
线性-80%浓度溶液的配制:精密量取线性贮备液4ml,置50ml量瓶中,用水稀释至刻度,摇匀。
[0090]
线性-50%浓度溶液的配制:精密量取线性贮备液2.5ml,置50ml量瓶中,用水稀释至刻度,摇匀。
[0091]
线性-定量限溶液的配制:精密量取线性贮备液1ml,置50ml量瓶中,用水稀释至刻度,摇匀。
[0092]
分别由不同实验人员在不同的仪器上进行线性与校正因子试验,检测条件如下:色谱柱:atlantis t3柱;流动相:以20mmol/l六氟磷酸钾缓冲液(用磷酸调ph至3.0)为流动相a,以乙腈为流动相b,梯度洗脱;流速:1.0ml/min;柱温:35℃;检测波长:215nm;进样量5μl。试验结果如下表4所示。
[0093]
表4线性范围及校正因子试验结果
[0094]
[0095][0096]
结果显示,各杂质在0.03%~0.30%范围内线性关系良好,相关系数r2≥0.999,校正因子在1.09~3.20之间,均可采用加校正因子的自身对照法计算。
[0097]
实施例5准确度试验
[0098]
准确度杂质储备液:精密量取各杂质贮备液5ml置同一100ml量瓶中,加水稀释至刻度,摇匀。
[0099]
定量限准确度溶液:取3-氨基-6-甲氧基哒嗪供试品约25mg,精密称定,置50ml量瓶中,加水超声使溶解,冷却至室温,精密加入准确度杂质对照品贮备液1ml,用水稀释至刻度,摇匀。
[0100]
定量限准确度对照溶液:精密量量取定量限准确度溶液2ml,置100ml量瓶中,用水稀释至刻度,摇匀;再精密量取该溶液5ml,置50ml量瓶中,用水稀释至刻度,摇匀。
[0101]
100%准确度溶液:取3-氨基-6-甲氧基哒嗪供试品约25mg,精密称定,置50ml量瓶中,加水超声使溶解,冷却至室温,精密加入准确度杂质对照品贮备液5ml,用水稀释至刻度,摇匀。
[0102]
100%准确度对照溶液:精密量量取100%准确度溶液2ml,置100ml量瓶中,用水稀释至刻度,摇匀;再精密量取该溶液5ml,置50ml量瓶中,用水稀释至刻度,摇匀。
[0103]
150%准确度溶液:取3-氨基-6-甲氧基哒嗪供试品约25mg,精密称定,置50ml量瓶中,加水超声使溶解,冷却至室温,精密加入准确度杂质对照品贮备液7.5ml,用水稀释至刻度,摇匀。
[0104]
150%准确度对照溶液:精密量量取150%准确度溶液2ml,置100ml量瓶中,用水稀释至刻度,摇匀;再精密量取该溶液5ml,置50ml量瓶中,用水稀释至刻度,摇匀。
[0105]
将样品进样检测,检测条件如下:色谱柱:atlantis t3柱;流动相:以20mmol/l六氟磷酸钾缓冲液(用磷酸调ph至3.0)为流动相a,以乙腈为流动相b,梯度洗脱;流速:1.0ml/min;柱温:35℃;检测波长:215nm;进样量5μl。通过测得值和理论加入量计算加标回收率,结果如下表5所示。
[0106]
表5加标回收率结果
[0107][0108]
上表结果显示,各杂质在不同浓度水平加标回收率均在94%~106%之间,且回收率rsd均小于5%,该方法准确度高。
[0109]
实施例6重复性和精密度试验
[0110]
由不同实验员配制相当于供试品浓度0.15%的杂质a和杂质b加标溶液作为测试溶液,配制方法如下:
[0111]
重复性(精密度)杂质对照品贮备液:精密量取杂a和杂质b贮备液各5ml,置100ml量瓶中,用水稀释至刻度,摇匀。
[0112]
重复性(精密度)溶液:取3-氨基-6-甲氧基哒嗪供试品约25mg,精密称定,置50ml量瓶中,加水超声使溶解,冷却至室温,分别精密加入重复性杂质对照品贮备液5ml,用水稀释至刻度,摇匀,平行配制6份。
[0113]
重复性(精密度)对照溶液:精密量重复性溶液2ml,置100ml量瓶中,用水稀释至刻度,摇匀,再精密量取该溶液5ml,置50ml量瓶中,用水稀释至刻度,摇匀。取6份重复性溶液,分别配制对照溶液。
[0114]
分别于不同日期、不同仪器上考察方法的重复性和精密度,检测条件如下:色谱柱:atlantis t3柱;流动相:以20mmol/l六氟磷酸钾缓冲液(用磷酸调ph至3.0)为流动相a,以乙腈为流动相b,梯度洗脱;流速:1.0ml/min;柱温:35℃;检测波长:215nm;进样量5μl。结果如表6所示。
[0115]
表6重复性与精密度试验结果
[0116][0117]
上表结果显示,两杂质的重复性rsd值分别为1.3%和0.5%,中间精密度rsd值分别为2.3%和1.8%,该方法重复性和精密度都较好。
[0118]
实施例7耐用性试验
[0119]
系统适用性溶液的配制:按照实施例1的配制方式配制系统适用性溶液。
[0120]
供试品溶液:精密称取3-氨基-6-甲氧基哒嗪供试品约25mg,置于50ml量瓶中,加水超声使溶解,再加水至刻度,摇匀;
[0121]
对照溶液:量取上述供试品溶液2ml,置100ml量瓶中,用水稀释至刻度,摇匀,得溶液i;量取溶液i 5ml,置50ml量瓶中,用水稀释至刻度,摇匀。
[0122]
改变色谱条件(柱温、流速、缓冲盐浓度、ph、检测波长、色谱柱),对系统适用性溶液、供试品溶液和对照溶液进样检测,记录结果,如下表7和表8所示。
[0123]
计算公式:
[0124][0125]
[0126]
总杂(%)=未知单杂之和(%)+已知杂质之和(%);
[0127]
其中a
杂质
—供试品溶液中各已知杂质的峰面积;a
对照
—对照溶液峰面积;f—各已知杂质校正因子。
[0128]
表7不同色谱条件下关键峰的分离度
[0129][0130]
表8不同色谱条件下检测结果
[0131][0132]
从上表7和8中的数据可知,色谱条件的微小变化对检测结果几乎无影响,该方法耐用性好。
[0133]
对比实施例1
[0134]
杂质a和杂质b系统适用性溶液的配制:准确称取杂质a、杂质b和3-氨基-6甲氧基哒嗪供试品适量,加水溶解并稀释至含杂质a0.75μg/ml、杂质b 0.3μg/ml、3-氨基-6甲氧基哒嗪0.5mg/ml的混合溶液。
[0135]
将该系统适用性溶液进样检测并记录结果,检测条件如下:色谱柱:xbridge shield rp18柱,3.5μm
×
4.6mm
×
150mm;流动相:以10mmol/l磷酸二氢钾和磷酸氢二钾溶液为流动相a,以乙腈为流动相b,梯度洗脱;流速:1.0ml/min;柱温:35℃;检测波长:215nm;进样体积:5μl,梯度程序如下:
[0136]
时间(min)流动相a(%)流动相b(%)0100010100025752525.011000351000
[0137]
色谱图如图4所示。
[0138]
结果表明在该色谱条件下,杂质b无保留,杂质a和主峰3-氨基-6甲氧基哒嗪分离不佳。
[0139]
对比实施例2
[0140]
杂质a和杂质b系统适用性溶液:准确称取杂质a、杂质b和3-氨基-6甲氧基哒嗪对照品适量,加水溶解并稀释至含杂质a0.75μg/ml、杂质b 0.3μg/ml、3-氨基-6甲氧基哒嗪0.5mg/ml的混合溶液。
[0141]
将该系统适用性溶液进样检测并记录结果,检测条件如下:色谱柱:xbridge shield rp18柱,3.5μm
×
4.6mm
×
150mm;流动相:以20mmol/l六氟磷酸钾溶液(磷酸调节ph至3.0)为流动相a,以乙腈为流动相b,梯度洗脱;流速:1.0ml/min;柱温:35℃;检测波长:215nm;进样体积:10μl,梯度程序如下:
[0142]
时间(min)流动相a(%)流动相b(%)0100010100025752525.011000351000
[0143]
色谱图如图5所示。
[0144]
结果表明在该色谱条件下,杂质a和主峰3-氨基-6甲氧基哒嗪虽分离良好,但杂质b无保留。
[0145]
对比实施例3
[0146]
系统适用性溶液:按照实施例1配制系统适用性溶液。
[0147]
将该系统适用性溶液进样检测并记录结果,检测条件如下:色谱柱:atlantis t3柱,3μm
×
4.6mm
×
150mm;流动相:以20mmol/l六氟磷酸钾缓冲液(用磷酸调ph至2.7)为流动相a,以乙腈为流动相b,梯度洗脱;流速:1.0ml/min;柱温:30℃;检测波长:215nm;进样体积:5μl,梯度程序如下:
[0148]
时间(min)流动相a(%)流动相b(%)010005100025752525.011000351000
[0149]
色谱图如图6所示。
[0150]
结果表明,在该色谱条件下,杂质e和杂质f未基线分离。
[0151]
对比实施例4
[0152]
系统适用性溶液:按照实施例1配制系统适用性溶液。
[0153]
将该系统适用性溶液进样检测并记录结果,检测条件如下:色谱柱:atlantis t3柱,3μm
×
4.6mm
×
150mm;流动相:以20mmol/l六氟磷酸钾缓冲液(用磷酸调ph至3.3)为流动相a,以乙腈为流动相b,梯度洗脱;流速:1.0ml/min;柱温:37℃;检测波长:215nm;进样体积:5μl,梯度程序如下:
[0154]
时间(min)流动相a(%)流动相b(%)0100010100025752525.011000351000
[0155]
色谱图如图7所示。
[0156]
结果表明,在该色谱条件下,杂质c和主峰3-氨基-6-甲氧基哒嗪分离不佳。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1