三化汤基准样品冻干粉指纹图谱的构建方法及其指纹图谱
1.本发明属于药物治疗控制领域,涉及一种中药经典名方三化汤的质量控制,具体涉及三化汤基准样品冻干粉指纹图谱的构建方法及其指纹图谱。
背景技术:
2.为贯彻落实《中华人民共和国中医药法》,推动来源于古代经典名方的中药复方制剂稳步发展,为人民群众健康提供更好保障,2018年4月16日国家中医药管理局会同国家药品监督管理局制定并发布《古代经典名方目录(第一批)》,为经典名方的研发带来了发展契机。
3.经典名方三化汤出自《素问病机气宜保命集》,现为国家公布的《古代经典名方目录(第一批)》的第55首。该方由大黄、枳实、厚朴和羌活组成,是治疗中风病之名方,具有通腑泄浊、升清降浊、调畅气机的作用,可有效改善缺血性脑水肿和脑血屏障通透性,多用于缺血性脑卒中的临床治疗。该方使用羌活以化风、厚朴和大黄以化滞、枳实以化痰,诸药合用,上能宣通脑之玄府,下能开通肠胃玄府,使全身津液畅通、气血调和,具有很好的临床应用前景。但由于中药复方本身成分非常复杂,并且成分之间相互影响,加大质量控制难度,目前,对三化汤成分分析的研究较少,多数为方中单味药的成分研究,不能反映三化汤的整体成分信息,亦无法为其质量提供控制与评价依据。
技术实现要素:
4.本发明的目的是解决现有技术的不足,提供三化汤基准样品冻干粉指纹图谱的构建方法及其指纹图谱,为方中大黄、枳实、厚朴、羌活提供全面的指纹图谱,该方法及其指纹图谱可以客观、全面、准确的评价三化汤基准样品冻干粉的质量,对控制三化汤的质量和保证临床疗效具有重要应用价值。
5.本发明通过以下技术方案实现:
6.本发明三化汤基准样品冻干粉指纹图谱的构建方法,包括基于三化汤基准样品冻干粉中大黄、枳实、厚朴药材的指纹图谱构建方法,以及基于三化汤基准样品冻干粉中羌活的指纹图谱构建方法,互为补充。
7.1.基于三化汤基准样品冻干粉中大黄、枳实、厚朴药材的指纹图谱构建方法,包括以下步骤:
8.s1、三化汤基准样品冻干粉供试品溶液的制备:
9.分别精密称取不同批次的三化汤基准样品冻干粉,置于具塞锥形瓶中,加甲醇溶液,超声提取,过0.45μm微孔滤膜,得供试品溶液;
10.s2、单一对照品溶液的制备:
11.分别精密称定没食子酸、原儿茶酸、柚皮苷、紫丁香苷、橙皮素、大黄素,置于容量瓶中,用乙醇定容至刻度;分别精密称定芦丁、紫花前胡苷、橙皮苷、新橙皮苷、槲皮素、芦荟大黄素、大黄素甲醚、山奈酚、和厚朴酚、厚朴酚、大黄酸和大黄酚,置于容量瓶中,用甲醇定
容至刻度;精密称定阿魏酸,置于容量瓶中,用热水定容至刻度,摇匀,制成单一对照品溶液;
12.s3、分别精密吸取s1供试品溶液和s2对照品溶液,注入高效液相色谱仪,记录色谱图;
13.s4、将s3中获得的三化汤基准样品冻干粉供试品溶液的色谱图导出,并导入中药色谱指纹图谱相似度评价系统2004a;选择不同批次三化汤基准样品冻干粉的色谱图中均存在的色谱峰作为共有峰;用平均值计算法生成三化汤基准样品冻干粉的对照指纹图谱r,导出不同批次三化汤基准样品冻干粉供试品溶液的色谱图的相似度结果表;并根据单一对照品溶液色谱图的保留时间标注对照指纹图谱中峰的化学成分,使用自动生成的对照指纹图谱r来生成共有色谱峰模式,分析计算获得多批三化汤基准样品冻干粉色谱图与共有色谱峰模式之间的相似度结果表并导出;根据相似度结果表及多批三化汤基准样品冻干粉色谱图多批三化汤基准样品冻干粉,确认结果的可靠性;
14.s5、将s3中获得的多批三化汤基准样品冻干粉的色谱图与单一对照品溶液的色谱图进行比对,指认出色谱图中1号峰为没食子酸、2号峰为原儿茶酸、3号峰为紫丁香苷、4号峰为阿魏酸、5号峰为芦丁、6号峰为紫花前胡苷、7号峰为柚皮苷、8号峰为橙皮苷、9号峰为新橙皮苷、10号峰为槲皮苷、11号峰为橙皮素、12号峰为山奈酚、13号峰为芦荟大黄素、14号峰为大黄酸、15号峰为和厚朴酚、16号峰为大黄素、17号峰为厚朴酚、18号峰为大黄酚、19号峰为大黄素甲醚。将分离度好、峰型好的一批三化汤基准样品冻干粉色谱图作为指纹图谱。
15.作为优选方案,s1中三化汤基准样品冻干粉供试品溶液制备方法为:取15批次的三化汤基准样品冻干粉1.25g,置250ml具塞锥形瓶中,加甲醇50ml,超声提取45min,过0.45μm微孔滤膜,得供试品溶液。
16.作为优选方案,s2中单一对照品溶液的制备:
17.分别精密称定没食子酸、原儿茶酸、柚皮苷、紫丁香苷、橙皮素、大黄素,置于容量瓶中,用乙醇定容至刻度;分别精密称定芦丁、紫花前胡苷、橙皮苷、新橙皮苷、槲皮素、芦荟大黄素、大黄素甲醚、山奈酚、和厚朴酚、厚朴酚、大黄酸和大黄酚,置于容量瓶中,用甲醇定容至刻度;精密称定阿魏酸,置于容量瓶中,用热水定容至刻度,摇匀,制成100μg/ml没食子酸、200μg/ml原儿茶酸、100μg/ml紫丁香苷、40μg/ml阿魏酸、100μg/ml芦丁、100μg/ml紫花前胡苷、100μg/ml柚皮苷、600μg/ml橙皮苷、100μg/ml新橙皮苷、100μg/ml槲皮素、100μg/ml橙皮素、100μg/ml山奈酚、100μg/ml芦荟大黄素、100μg/ml大黄酸、100μg/ml和厚朴酚、100μg/ml大黄素、100μg/ml厚朴酚、100μg/ml大黄酚和100μg/ml大黄素甲醚的单一对照品溶液。
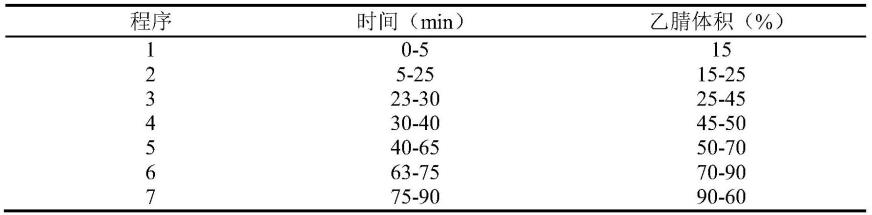
18.作为优选方案,s3中液相色谱条件为:色谱柱:kromasil 100-5-c18(250mm
×
4.6mm,5μm)色谱柱,流动相:乙腈和0.1%磷酸水溶液,梯度洗脱,紫外可见吸收检测器多波长切换检测,检测波长:0~35min,280nm;35~45min,254nm;45~58min,310nm;58~90min,254nm;柱温30℃,流速1.0ml/min,进样体积:20μl,洗脱程序如下表:
[0019][0020]
2.基于三化汤基准样品冻干粉中大黄、枳实、厚朴药材的指纹图谱构建方法的优化:
[0021]
s1、在样品溶液制备方面的优化
[0022]
本发明通过对不同提取方法(超声、回流、浸渍)及不同提取溶剂(二氯甲烷、三氯甲烷、石油醚、甲醇、50%甲醇水溶液、70%甲醇水溶液、水)进行实验比较,结果发现超声提取所得的色谱图成分比较全面,分离度良好,故采用超声提取的方法;对提取溶剂的考察中发现甲醇提取物色谱图信息量最多,成分含量最高;所以选用甲醇进行提取。
[0023]
s2、在色谱条件方面进行优化
[0024]
本发明采用紫外可见吸收检测器对检测波长进行考察,提取254nm、280nm、310nm处的色谱图,发现检测波长条件为:0~35min,280nm;35~45min,254nm;45~58min,310nm;58~90min,254nm时,色谱图所包含的信息量最全面且基线平稳,故选此方法为检测波长条件。
[0025]
本发明对流速(0.6ml/min、0.8ml/min、1.0ml/min)进行筛选,发现流速影响较小,且各个物质的分离效果均良好,保持1.0ml/min的流速。
[0026]
本发明比较了甲醇-水,乙腈-水,甲醇-乙腈-水、甲醇-0.1%磷酸水、乙腈-0.1%磷酸水、乙腈-0.05%磷酸水、乙腈-0.2%磷酸水、乙腈-0.1%甲酸,乙腈-0.1%冰醋酸、乙腈-三乙胺水多个不同洗脱系统在不同梯度下的洗脱效果。结果发现以乙腈和0.1%磷酸水为流动相时,三化汤中各成分分离效果较好,故最终选定以乙腈和0.1%磷酸水为流动相。
[0027]
在确定最佳流动相组成后,本发明通过大量实验筛选最佳的梯度洗脱程序,实验发现,当采用0~5min乙腈体积15%;5~25min乙腈体积15%~25%;23~30min乙腈体积25%~45%;30~40min乙腈体积45%~50%;40~65min乙腈体积50%~70%;63~75min乙腈体积70%~90%;75~90min乙腈体积90%~60%时可实现指纹图谱中各色谱峰的良好分离度。
[0028]
3.基于三化汤基准样品冻干粉中羌活药材的指纹图谱构建方法,包括以下步骤:
[0029]
ss1、三化汤基准样品冻干粉供试品溶液的制备:
[0030]
分别精密称取不同批次的三化汤基准样品冻干粉,置于具塞锥形瓶中,加甲醇溶液,超声提取,过0.45μm微孔滤膜,得供试品溶液;
[0031]
ss2、单一对照品溶液的制备:
[0032]
分别精密称定羌活醇、阿魏酸苯乙醇酯、异欧前胡素和镰叶芹二醇,分别置于容量瓶中,用甲醇定容至刻度,摇匀,制成单一对照品溶液;
[0033]
ss3、分别精密吸取ss1中供试品溶液和ss2中单一对照品溶液,注入高效液相色谱仪,记录色谱图;
[0034]
ss4、将ss3中获得的三化汤基准样品冻干粉供试品溶液的色谱图导出,并导入中
药色谱指纹图谱相似度评价系统2004a;选择不同批次三化汤基准样品冻干粉的色谱图中均存在的色谱峰作为共有峰;用平均值计算法生成三化汤基准样品冻干粉的对照指纹图谱r,导出不同批次三化汤基准样品冻干粉供试品溶液的色谱图的相似度结果表;并根据单一对照品溶液色谱图的保留时间标注对照指纹图谱中峰的化学成分,使用自动生成的对照指纹图谱r来生成共有色谱峰模式,分析计算获得多批三化汤基准样品冻干粉色谱图与共有色谱峰模式之间的相似度结果表并导出;根据相似度结果表及多批三化汤基准样品冻干粉色谱图多批三化汤基准样品冻干粉,确认结果的可靠性;
[0035]
ss5、将ss3中获得的多批三化汤基准样品冻干粉色谱图和单一对照品色谱图进行比对,指认出色谱图中1号峰为羌活醇、2号峰为阿魏酸苯乙醇酯、3号峰为异欧前胡素、4号峰为镰叶芹二醇,将分离度好、峰型好的一批三化汤基准样品冻干粉色谱图作为指纹图谱。
[0036]
作为优选方案,ss1中三化汤基准样品冻干粉供试品溶液制备方法为:15批次的三化汤基准样品冻干粉1.25g,置250ml具塞锥形瓶中,加甲醇50ml,超声提取45min,过0.45μm微孔滤膜,得供试品溶液。
[0037]
作为优选方案,ss2中单一对照品溶液的制备:分别取精密称定羌活醇、阿魏酸苯乙醇酯、异欧前胡素和镰叶芹二醇对照品,置于容量瓶中,用甲醇定容至刻度,摇匀,制成60μg/ml羌活醇、30μg/ml阿魏酸苯乙醇酯、100μg/ml异欧前胡素、20μg/ml镰叶芹二醇的单一对照品溶液。
[0038]
作为优选方案,液相色谱条件为:色谱柱:kromasil 100-5-c18(250mm
×
4.6mm,5μm)色谱柱,流动相:乙腈和0.1%磷酸水溶液,梯度洗脱,检测波长:246nm;柱温25℃;流速1.0ml/min;进样体积:20μl,洗脱程序如下表:
[0039][0040]
4.基于三化汤基准样品冻干粉中羌活药材的指纹图谱构建方法的优化:
[0041]
ss1、在样品溶液制备方面的优化
[0042]
本发明通过对不同提取方法(超声、回流、浸渍)及不同提取溶剂(二氯甲烷、三氯甲烷、石油醚、甲醇、50%甲醇水溶液、70%甲醇水溶液、水)进行实验比较,结果发现超声提取所得的色谱图成分比较全面,分离度良好,故采用超声提取的方法;对提取溶剂的考察中发现甲醇提取物色谱图信息量最多,成分含量最高;所以选用甲醇进行提取。
[0043]
ss1、在色谱条件方面进行优化
[0044]
本发明采用紫外可见吸收检测器对检测波长进行考察,提取246nm、254nm、280nm、284nm、300nm处的色谱图,发现检测波长为246nm时,色谱图所包含的信息量最全面且基线平稳,故选246nm为检测波长;
[0045]
本发明对流速(0.5ml/min、0.6ml/min、0.7ml/min、0.8ml/min、1.0ml/min)进行筛选,发现流速影响较小,保持1.0ml/min的流速。
[0046]
本发明对柱温(25℃、28℃、30℃)进行筛选,发现柱温影响较小,保持25℃的柱温。
[0047]
本发明比较了甲醇-水,乙腈-水,乙腈-0.1%甲酸,乙腈和0.05%磷酸水,乙腈-0.1%磷酸水5个不同洗脱系统在不同梯度下的洗脱效果。结果发现以乙腈和0.1%磷酸水
为流动相时,三化汤中各成分分离效果较好,故最终选定以乙腈和0.1%磷酸水为流动相。
[0048]
在确定最佳流动性组成后,本发明通过大量实验筛选最佳的洗脱程序,实验发现,当采用0~6min乙腈体积48%;6~12min乙腈体积53%;12~40min乙腈体积73%时可实现指纹图谱中各色谱峰的良好分离度。
[0049]
本发明三化汤基准样品冻干粉由酒大黄、姜厚朴、麸炒枳实、去芦羌活以上四味药材,置于煎药锅中,加适量纯化水浸泡一定时间,煎煮两次,喷雾冷冻干燥制成。
[0050]
与现有技术相比,本发明具有如下的有益效果:
[0051]
本发明在利用多波长切换,同法建立三化汤基准样品冻干粉中大黄、枳实、厚朴三味药材指纹图谱基础上,另建立新的指纹图谱方法,提供了基准样品中羌活的检测手段。两指纹图谱互为补充,构成一套完整的指纹图谱,为方中大黄、枳实、厚朴、羌活提供全面的指纹图谱。经多次实验验证表明,本发明提供的三化汤基准样品冻干粉的指纹图谱构建方法,各色谱峰分离较好,基线平稳,峰型好,具有全面客观、稳定性好、精密度高、重现性好等优点,能全面的反应其中所含化学成分的种类与数量,可以全面、客观、准确的检测和评价三化汤基准样品冻干粉的质量,为保证临床三化汤基准样品冻干粉疗效具有重要意义。
[0052]
进一步的,在供试品溶液制备方面,本发明通过对不同提取方法(超声、回流、浸渍)及不同提取溶剂(二氯甲烷、三氯甲烷、石油醚、甲醇、50%甲醇水溶液、70%甲醇水溶液、水)进行实验比较,结果发现超声提取所得的色谱图成分比较全面,分离度良好,故采用超声提取的方法;对提取溶剂的考察中发现甲醇提取物色谱图信息量最多,成分含量最高;所以选用甲醇进行提取。
[0053]
进一步的,本发明比较了s3中多个不同洗脱系统在不同梯度下的洗脱效果,结果发现以乙腈和0.1%磷酸水为流动相时,三化汤中各成分分离效果较好,故最终选定以乙腈和0.1%磷酸水为流动相,在确定最佳流动相组成后,本发明通过大量实验筛选最佳的梯度洗脱程序,实验发现,当采用0~5min乙腈体积15%;5~25min乙腈体积15%~25%;23~30min乙腈体积25%~45%;30~40min乙腈体积45%~50%;40~65min乙腈体积50%~70%;63~75min乙腈体积70%~90%;75~90min乙腈体积90%~60%时可实现指纹图谱中各色谱峰的良好分离度。
[0054]
进一步的,本发明比较了ss3中多个不同洗脱系统在不同梯度下的洗脱效果,结果发现以乙腈和0.1%磷酸水为流动相时,三化汤中各成分分离效果较好,故最终选定以乙腈和0.1%磷酸水为流动相,在确定最佳流动性组成后,本发明通过大量实验筛选最佳的洗脱程序,实验发现,当采用0~6min乙腈体积48%;6~12min乙腈体积53%;12~40min乙腈体积73%时可实现指纹图谱中各色谱峰的良好分离度。
[0055]
用本发明所提供的方法所建立的三化汤基准样品冻干粉的指纹图谱,能有效地表征三化汤基准样品冻干粉的质量,能客观体现各个构成指纹特征峰的前后顺序和相互关系,注重整体面貌特征,既可避免因测定个别化学成分而判定三化汤基准样品冻干粉质量的片面性,又可减少为质量达标而人为处理的可能性。
附图说明
[0056]
图1为本发明基于三化汤基准样品冻干粉中大黄、枳实、厚朴药材19个峰的对照指纹图谱。
[0057]
图2为本发明没食子酸对照品溶液的色谱图。
[0058]
图3为本发明原儿茶酸对照品溶液的色谱图。
[0059]
图4为本发明紫丁香苷对照品溶液的色谱图。
[0060]
图5为本发明阿魏酸对照品溶液的色谱图。
[0061]
图6为本发明芦丁对照品溶液的色谱图。
[0062]
图7为本发明紫花前胡苷对照品溶液的色谱图。
[0063]
图8为本发明柚皮苷对照品溶液的色谱图。
[0064]
图9为本发明橙皮苷对照品溶液的色谱图。
[0065]
图10为本发明新橙皮苷对照品溶液的色谱图。
[0066]
图11为本发明槲皮素对照品溶液的色谱图。
[0067]
图12为本发明橙皮素对照品溶液的色谱图。
[0068]
图13为本发明山奈酚对照品溶液的色谱图。
[0069]
图14为本发明和厚朴酚对照品溶液的色谱图。
[0070]
图15为本发明厚朴酚对照品的溶液色谱图。
[0071]
图16为本发明大黄混标(芦荟大黄素、大黄酸、大黄素、大黄酚、大黄素甲醚对照品)的色谱图。
[0072]
图17为本发明没食子酸对照品溶液的质谱图。
[0073]
图18为本发明原儿茶酸对照品溶液的质谱图。
[0074]
图19为本发明阿魏酸对照品溶液的质谱图。
[0075]
图20为本发明紫丁香苷对照品溶液的质谱图。
[0076]
图21为本发明芦丁、橙皮苷、新橙皮苷对照品溶液的质谱图。
[0077]
图22为本发明柚皮苷对照品溶液的质谱图。
[0078]
图23为本发明紫花前胡苷对照品溶液的质谱图。
[0079]
图24为本发明槲皮素、橙皮素对照品溶液的质谱图。
[0080]
图25为本发明山奈酚对照品溶液的质谱图。
[0081]
图26为本发明芦荟大黄素、大黄素对照品溶液的质谱图。
[0082]
图27为本发明大黄酸对照品溶液的质谱图。
[0083]
图28为本发明和厚朴酚、厚朴酚对照品溶液的质谱图。
[0084]
图29为本发明大黄酚对照品溶液的质谱图。
[0085]
图30为本发明大黄素甲醚对照品溶液的质谱图。
[0086]
图31为本发明基于三化汤基准样品冻干粉中大黄、枳实、厚朴药材的15批次供试品溶液色谱图。
[0087]
图32为本发明基于三化汤基准样品冻干粉中羌活药材的对照指纹图谱。
[0088]
图33为本发明羌活醇对照品溶液的色谱图。
[0089]
图34为本发明阿魏酸苯乙醇酯对照品溶液的色谱图。
[0090]
图35为本发明异欧前胡素对照品溶液的色谱图。
[0091]
图36为本发明镰叶芹二醇对照品溶液的色谱图。
[0092]
图37为本发明基于三化汤基准样品冻干粉中羌活药材的15批次供试品色谱图。
具体实施方式
[0093]
下面将结合实施例对本发明的实施方案进行详细描述,实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。
[0094]
实施例用到的仪器与试剂如下:
[0095]
实验器材
[0096]
1.仪器
[0097]
岛津仪器(中国苏州)股份有限公司lc-20(uv)高效液相色谱仪,包括lc-20送液单元,sil-20自动进样器,spd-20紫外可见双波长检测器和cto-20储液盒一体型柱温箱,ym-100s型数控超声波清洗器(深圳市方奥微电子有限公司),6200系列tof/6500系列质谱仪(agilent),ja2603b电子分析天平(上海天美天平仪器有限公司),le204e/02电子天平(梅特勒-托利多仪器有限公司),jcs-600电子天平(凯丰集团有限公司)。
[0098]
2.药品与试剂
[0099]
三化汤基准样品冻干粉样品为自制,取酒大黄、姜厚朴、麸炒枳实、去芦羌活以上四味药材,置于煎药锅中,浸泡一定时间,煎煮两次,喷雾冷冻干燥制成。
[0100]
对照品:没食子酸对照品(批号110831-201906);原儿茶酸对照品(批号110809-201906);紫丁香苷对照品(批号111574-201605);阿魏酸对照品(批号110773-201915);芦丁对照品(批号100080-202012);紫花前胡苷对照品(批号111821-201604);柚皮苷对照品(批号110722-202116);橙皮苷对照品(批号110721-202019);新橙皮苷对照品(批号111857-201804);槲皮素对照品(批号100081-201610);山奈酚对照品(批号110861-202013);芦荟大黄素对照品(批号110795-202011);大黄酸对照品(批号110757-201607);羌活醇对照品(批号111820-201705);大黄素对照品(批号110756-201913);异欧前胡素对照品(批号110827-201812);大黄酚对照品(批号110796-201922);大黄素甲醚对照品(批号110758-201817);和厚朴酚对照品(批号110730-201915);厚朴酚(批号110729-202015);以上对照品均购自中国食品药品检定研究所;橙皮素对照品(批号h23331)购自上海吉至生化科技有限公司;阿魏酸苯乙醇酯对照品(批号p11329)购自北京谨明生物科技有限公司;镰叶芹二醇对照品(批号f74680)购自上海吉至生化科技有限公司;甲醇(分析纯);磷酸(分析纯);乙腈(色谱纯)。
[0101]
实施例1一种基于三化汤基准样品冻干粉中大黄、枳实、厚朴药材的指纹图谱构建方法,包括以下步骤:
[0102]
s1、三化汤基准样品冻干粉供试品溶液的制备:
[0103]
取15批次的三化汤基准样品冻干粉1.25g置250ml具塞锥形瓶中,加甲醇50ml,超声45min,过0.45μm微孔滤膜,得供试品溶液;
[0104]
s2、单一对照品溶液的制备:
[0105]
分别精密称定没食子酸、原儿茶酸、柚皮苷、紫丁香苷、橙皮素、大黄素,置于容量瓶中,用乙醇定容至刻度;分别精密称定芦丁、紫花前胡苷、橙皮苷、新橙皮苷、槲皮素、芦荟大黄素、大黄素甲醚、山奈酚、和厚朴酚、厚朴酚、大黄酸和大黄酚,置于容量瓶中,用甲醇定容至刻度;精密称定阿魏酸,置于容量瓶中,用热水定容至刻度,摇匀,制成含100μg/ml没食子酸、200μg/ml原儿茶酸、100μg/ml紫丁香苷、40μg/ml阿魏酸、100μg/ml芦丁、100μg/ml紫
花前胡苷、100μg/ml柚皮苷、600μg/ml橙皮苷、100μg/ml新橙皮苷、100μg/ml槲皮素、100μg/ml橙皮素、100μg/ml山奈酚、100μg/ml芦荟大黄素、100μg/ml大黄酸、100μg/ml和厚朴酚、100μg/ml大黄素、100μg/ml厚朴酚、100μg/ml大黄酚和100μg/ml大黄素甲醚的单一对照品溶液;
[0106]
s3、分别精密吸取15批三化汤基准样品冻干粉供试品溶液和单一对照品溶液,注入高效液相色谱仪,记录色谱图;液相色谱条件为:色谱柱:kromasil 100-5-c18(250mm
×
4.6mm,5μm)色谱柱,流动相:乙腈和0.1%磷酸水溶液,梯度洗脱,紫外可见吸收检测器多波长切换,检测波长:0~35min,280nm;35~45min,254nm;45~58min,310nm;58~90min,254nm;柱温30℃,流速1.0ml/min,进样体积:20μl,洗脱程序如下表:
[0107][0108]
s4、将s3中获得的15批三化汤基准样品冻干粉供试品溶液的色谱图导出,并导入中药色谱指纹图谱相似度评价系统2004a;选择15批次三化汤基准样品冻干粉的色谱图中均存在的色谱峰作为共有峰;用平均值计算法生成三化汤基准样品冻干粉的对照指纹图谱r,导出不同批次三化汤基准样品冻干粉供试品溶液的色谱图的相似度结果表;并根据单一对照品溶液色谱图的保留时间标注对照指纹图谱中峰的化学成分,使用自动生成的对照指纹图谱r来生成共有色谱峰模式,分析计算获得15批三化汤基准样品冻干粉色谱图与共有色谱峰模式之间的相似度结果表并导出,如表1;结果1批三化汤基准样品冻干粉中有19个共有峰,15批次供试品的色谱图及对照指纹图谱r见图31所示;根据相似度结果表1及图31可以看出,15批三化汤基准样品冻干粉色谱图与共有色谱峰模式之间具有相对很好的相似性,因此确认结果的可靠性。
[0109]
表1各批次样品与共有色谱峰模式间的相似度
[0110][0111]
s5、s3中获得的三化汤基准样品冻干粉色谱图和单一对照品色谱图(图2~图16)
进行比对,结合图17-图30,指认主要成分,对比出三化汤基准样品冻干粉中1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19号色谱峰分别为:没食子酸(保留时间2.958min)、原儿茶酸(保留时间5.825min)、紫丁香苷(保留时间7.365min)、阿魏酸(保留时间16.133min)、芦丁(保留时间17.086min)、紫花前胡苷(保留时间18.751min)、柚皮苷(保留时间22.119min)、橙皮苷(保留时间23.681min)、新橙皮苷(保留时间21.786min)、槲皮素(保留时间33.973min)、橙皮素(保留时间35.869min)、山奈酚(保留时间37.492min)、芦荟大黄素(保留时间40.01min)、大黄酸(保留时间41.345min)、和厚朴酚(保留时间46.27min)、大黄素(保留时间48.329min)、厚朴酚(保留时间51.546min)、大黄酚(保留时间59.867min)、大黄素甲醚(保留时间70.385min)。将分离度好、峰型好的一批三化汤基准样品冻干粉色谱图作为最终的指纹图谱,见图1。
[0112]
实施例2基于三化汤基准样品冻干粉中大黄、枳实、厚朴药材的指纹图谱构建方法的方法学研究:
[0113]
s1、精密度研究
[0114]
取实施例1方法制备的柚皮苷对照品溶液,按照实施例1的检测方法分析,平行进样6次,进样量为20μl,通过分析峰面积及保留时间并计算rsd值,结果见表2,结果表明该设备的平行进样精密性良好。
[0115]
表2柚皮苷精密度研究峰面积和保留时间
[0116][0117]
s2、稳定性研究
[0118]
取三化汤基准样品冻干粉1.25g按实施例1方法制备供试品溶液,按照实施例1的检测方法分析,采用0、2、6、12、18、24h不同时间进样分析,进样量为20μl,以没食子酸、原儿茶酸、紫丁香苷、阿魏酸、芦丁、紫花前胡苷、柚皮苷、橙皮苷、新橙皮苷、槲皮素、橙皮素、山奈酚、芦荟大黄素、大黄酸、和厚朴酚、大黄素、厚朴酚、大黄酚和大黄素甲醚为参照峰,通过分析样品hplc色谱图的共有峰的峰面积及保留时间并计算rsd值,结果见表3,表明三化汤供试品溶液在24h之内的色谱峰几乎没有变化,稳定性好。
[0119]
表3稳定性研究峰面积和保留时间
[0120]
[0121][0122][0123]
s3、重复性研究
[0124]
按上述供试品溶液方法制备六批样品溶液,参照实施例1的色谱条件,进样量为20μl,以没食子酸、原儿茶酸、紫丁香苷、阿魏酸、芦丁、紫花前胡苷、柚皮苷、橙皮苷、新橙皮苷、槲皮素、橙皮素、山奈酚、芦荟大黄素、大黄酸、和厚朴酚、大黄素、厚朴酚、大黄酚和大黄
素甲醚为参照峰,通过分析样品hplc色谱图的共有峰的峰面积及保留时间并计算rsd值,结果见表4,结果表明样品色谱峰重现性好,该方法的重复性良好。
[0125]
表4重复性研究峰面积和保留时间
[0126][0127][0128]
实施例3一种基于三化汤基准样品冻干粉中羌活药材的指纹图谱检测方法,包括以下步骤:
[0129]
ss1、三化汤基准样品冻干粉供试品溶液的制备:
[0130]
取15批次的三化汤基准样品冻干粉1.25g置250ml具塞锥形瓶中,加甲醇,超声提取,过0.45μm微孔滤膜,得供试品溶液;
[0131]
ss2、单一对照品溶液的制备:
[0132]
分别精密称定羌活醇、阿魏酸苯乙醇酯、异欧前胡素和镰叶芹二醇,置于容量瓶中,用甲醇定容至刻度,摇匀,制成含60μg/ml羌活醇、30μg/ml阿魏酸苯乙醇酯、100μg/ml异欧前胡素、20μg/ml镰叶芹二醇的单一对照品溶液;
[0133]
ss3、分别精密吸取15批三化汤基准样品冻干粉供试品溶液和单一对照品溶液,注
入高效液相色谱仪,记录色谱图;液相色谱条件为:色谱柱:kromasil 100-5-c18(250mm
×
4.6mm,5μm)色谱柱,流动相:乙腈和0.1%磷酸水溶液,程序洗脱,检测波长:246nm;柱温25℃;流速1.0ml/min;进样体积:20μl,洗脱程序如下表:
[0134][0135]
ss4、将ss3中获得的15批三化汤基准样品冻干粉供试品溶液的色谱图导出,并导入中药色谱指纹图谱相似度评价系统2004a;选择15批次三化汤基准样品冻干粉的色谱图中均存在的色谱峰作为共有峰;用平均值计算法生成三化汤基准样品冻干粉的对照指纹图谱,使用自动生成的对照指纹图谱r来生成共有色谱峰模式,分析计算获得15批三化汤基准样品冻干粉色谱图与共有色谱峰模式之间的相似度结果表并导出,如表5;结果1批三化汤基准样品冻干粉中有4个共有峰,15批次供试品的色谱图及生成的对照指纹图谱r见图37所示;根据相似度结果表及图37可以看出,15批三化汤基准样品冻干粉色谱图与共有色谱峰模式之间具有相对很好的相似性,因此确认结果的可靠性。
[0136]
表5各批次样品与共有模式间的相似度
[0137][0138][0139]
ss5、将ss3中获得的15批次三化汤基准样品冻干粉色谱图和单一对照品色谱图(图33~图36)进行比对,指认主要成分,对比出三化汤基准样品冻干粉中1、2、3、4号色谱峰分别为:羌活醇(保留时间14.132min)、阿魏酸苯乙醇酯(保留时间15.983min)、异欧前胡素(保留时间18.287min)、镰叶芹二醇(保留时间25.595min)。将分离度好、峰型好的一批三化汤基准样品冻干粉色谱图作为最终的指纹图谱,见图32。
[0140]
实施例4基于三化汤基准样品冻干粉中羌活药材的指纹图谱构建方法的方法学研究:
[0141]
s1、精密度研究
[0142]
取按实施例2方法制备的异欧前胡素标准品,按照实施例2的检测方法分析,平行进样6次,进样量为20μl,通过分析hplc的峰面积及保留时间并计算rsd值,结果见表6结果表明该设备的平行进样精密性良好。
[0143]
表6异欧前胡素精密度研究峰面积和保留时间
[0144][0145]
s2、稳定性研究
[0146]
取三化汤基准样品冻干粉1.25g按实施例2方法制备的样品为供试品溶液,按照实施例2的检测方法分析,采用0、2、6、12、18、24h不同时间进样分析,进样量为20μl,以羌活醇、阿魏酸苯乙醇酯、异欧前胡素和镰叶芹二醇为参照峰,通过分析样品hplc指纹图谱的共有峰的峰面积及保留时间并计算rsd值,结果见表7,表明三化汤供试品溶液在24h之内的色谱峰几乎没有变化,稳定性好。
[0147]
表7稳定性研究峰面积和保留时间
[0148][0149][0150]
s3、重复性研究
[0151]
按上述供试品溶液方法制备六批样品溶液,参照实施例2的色谱条件,进样量为20μl,以羌活醇、阿魏酸苯乙醇酯、异欧前胡素和镰叶芹二醇为参照峰,通过分析样品hplc指纹图谱的共有峰的峰面积及保留时间并计算rsd值,结果见表8,结果表明样品色谱峰重现性好,该方法的重复性良好。
[0152]
表8重复性研究峰面积和保留时间
[0153][0154]
以上实验结果表明,本发明提供的三化汤基准样品冻干粉的指纹图谱构建方法,均具有稳定性好,精密度高,重复性好的特点,能全面客观评价三化汤的质量,为临床疗效提供质量保证。
[0155]
以上实施例仅为本发明的示例性实施例,不用于限制本发明,本发明的保护范围由权利要求书限定。本领域技术人员可以在本发明的实质和保护范围内,对本发明做出各种修改或等同替换,这种修改或等同替换也应视为落在本发明的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1