一种去除单萜类成分的艾草重油成分的检测方法
1.本发明属于轻工业香料检测方法领域,具体涉及一种去除单萜类成分的艾草重油成分的检测方法。
背景技术:
2.艾草是我国日常保健养生主要的植物,其药用历史悠久,位列于我国《药典》中药材目录中。艾草具有多种功效,如通经活络、抗炎、抑菌、止血止痛和抗病毒等生物学活性。多重的生物学功效来源于艾草中的化学成分,艾草含有萜类物质、黄酮类、鞣质类、多糖等成分。其中萜类成分是艾草主要的一类物质成分,主要有单萜类成分、倍半萜类成分和三萜类成分。现代研究证明单萜烯类具有杀菌、止痛、兴奋神经和一定程度的皮肤刺激,且单萜类物质容易挥发和氧化。而倍半萜烯类有舒缓、镇静、抗炎的功效,相对单萜类成分稳定性更好,留香更持久。
3.目前常规的水蒸馏提取、有机试剂萃取等提取艾叶油的方式获得的艾草油主要单萜类成分含量最高,因此造成艾草油容易挥发和氧化。在存储和使用周期上具有一定的局限性。
4.为了保持艾草油的特殊香味,降低皮肤刺激性、延长产品使用期限,使得艾草油在抗炎、舒缓镇静的作用更好,可以通过更为精细和先进的提取方式获得高含量倍半萜物质的艾草油,并且建立特征性成分的快速检测方法是精细化提升和控制艾草油品质的关键。
5.在我们的前期工艺研究过程中发现,艾草经超临界获得浸膏后,再经过分子蒸馏工艺,在一定压力及挥发温度下,利用浸膏的挥发成分中轻、重分子的分子自由程不同,在给予冷凝后,将轻组分分子直接冷凝排出,而重组分则因达不到冷凝板而掉落排出。根据该原理分离艾草浸膏中的轻组分单萜类和重组分倍半萜类,使得大部分单萜及少量沸点较低的倍半萜烯作为轻组分流出,而大部分重组分倍半萜类物质作为重组分。为了使得工艺结果数据可视化,工艺成品品质评价有所依据,检测反馈优化生产工艺结果,同时也可以作为艾草重油品质鉴定方法,因此需要建立一种艾草重油的检测方法。
6.超临界分子蒸馏工艺中沸点较高、分子自由程较大的这一段重油中部分成分(如十氢二甲基甲乙烯基苯酚neointermedeol)的检测方法是采用gc或者gcms定性分析的方法进行测定,无法进行准确定量,需采用定量离子和定性离子捕获的选择离子检测模式进行检测。例如,中国专利申请cn 112034084 a公开了一种艾油中挥发性成分的检测方法及其应用,涉及挥发性成分检测技术领域。该检测方法通过气相色谱-质谱联用检测的方法对艾油中挥发性成分进行检测,利用谱库检索的方法对全扫描“scan”模式下的色谱-质谱图进行检索定性,得到全部挥发性成分的定性数据;同时,利用分段扫描的方法对主要挥发性物质在“sim”模式下色谱-质谱图的峰面积进行分析,进而利用外标法计算得到各主要挥发性物质的含量。该方法针对艾油中单萜类挥发性成分进行检测,具有简单准确,专属性、实用性强的特点。但是该检测方法针对性极强,并不适用于艾草重油中主要成分的检测,因此本发明建立了去除单萜类成分的艾草重油的检测方法。
技术实现要素:
7.为了解决现有技术中存在的问题,本发明的目的在于提供去除单萜类成分的艾草重油成分的检测方法,本发明所述的gcms方法对艾草重油中的主要成分进行含量测定方法具有较高的准确度和精密度,且稳定性和重复性好,能准确、快速地进行定量分析,方法可行性,结果可靠。
8.本发明的技术方案是:
9.一种去除单萜类成分的艾草重油成分的检测方法,所述检测方法为气相色谱-质谱仪测定方法,包括以下步骤:
10.s1:将待测物标准品、c9-c40正构烷烃标准品和待测样品用甲醇或正己烷稀释制成溶液,其中,所述待测样品是去除单萜类成分的艾草重油;
11.s2:分别将待测物标准品溶液和待测样品溶液以相同的采集方法分别注入气相色谱-质谱联用仪中,同时进行scan全扫描+sim选择离子检测,得到待测物标准品溶液和待测样品溶液中化合物的定量离子及定性离子、离子流图eic以及总离子流图tic;将c9-c40正构烷烃标准品溶液以相同的采集方法注入气相色谱-质谱联用仪中,得c9-c40正构烷烃标准品溶液的总离子流图tic;
12.s3:定性分析:将c9-c40正构烷烃标准品溶液的总离子流图tic导入定性分析软件中,使用nist库对数据进行匹配,识别并获得c9-c40正构烷烃的保留时间;将待测样品采集的总离子流图导入分析软件中,采用解卷积计算方法,筛选与谱库匹配度>70%,获得定性分析检索化合物,根据各化合物的保留时间与正构烷烃的保留时间比值计算化合物的保留指数,对比化合物的标准保留指数与谱库检索的匹配度,获得艾草重油定性分析数据结果;
13.s4:定量分析:根据sim选择离子检测数据结果,使用定量分析软件,对质谱图进行分析,并分别对选择的定量离子、定性离子进行积分,获取选定待测物标准品溶液与待测样品响应比值、标准品溶液浓度,计算出待测物的浓度。
14.进一步地,所述的去除单萜类成分的艾草重油成分的检测方法,步骤s2所述气相色谱-质谱联用仪的色谱条件如下:
15.色谱柱:5%苯基甲基聚硅氧烷、100%聚二甲基硅氧烷、5%二苯基1%乙烯基二甲基聚硅氧烷、50%二苯基-50%二甲基聚硅氧烷、14%氰丙基苯基二甲基聚硅氧烷、50%氰丙基苯基二甲基聚硅氧烷、聚乙二醇、100%甲基聚硅氧烷、5%苯基聚硅氧烷或1%乙烯基5%苯基甲基聚硅氧烷为固定相的色谱柱;
16.升温程序:60℃-75℃保持2-5min,以2-5℃/min升至120℃-170℃保持2min-5min,以2-5℃/min升至240℃-250℃保持2min-5min,以10℃/min升至290℃保持1min;(升温程序是色谱分离条件的重要组成部分。艾草重油中各组分在色谱柱确定后(固定相确定),为保证一定的分离度,提高质谱测定后样品检索准确定的必要前提。不同的色谱柱的组分保留时间和分离度不同,而升温程序是随色谱柱和样品中各组分不同而变化的。)
17.进样口温度:240℃;
18.分流比:30:1~100:1;
19.所述气相色谱的载气为氮气、氦气、氢气,流速为0.8-1.5ml/min。优选地,氮气、氦气、氢气采用高纯氮气、氦气、氢气,纯度均大于99.995%。
20.本发明所检测的去除单萜类成分的艾草重油成分在100-200℃之间可以挥发,而
且所述艾草重油成分中的各组分结构很相近,因此需要使各组分的分离开,否则会影响最终的质量数选择、判断;然而,目前现有技术中的仪器无法精准做到升温程序中升温速率为0.2-0.5℃/分钟,从而使艾草重油成分中的结构相近各组分进行分离;而本发明所设定的升温程序能使所检测的去除单萜类成分的艾草重油成分在通过30m的色谱柱时,结构相近的组分能有一定的分离,使得各主要成分的质量数更易选择、判断。
21.更进一步地,所述气相色谱-质谱联用仪的最佳色谱条件如下:
22.色谱柱:5%苯基甲基聚硅氧烷为固定相的色谱柱,柱长30m;
23.升温程序:70℃保持2min,以5℃/min升至150℃保持2min,以3℃/min升至250℃保持2min,以10℃/min升至290℃保持1min;
24.进样口温度:240℃;
25.分流比:50:1;
26.所述气相色谱的载气为氦气,氦气纯度大于99.995%;氦气流速为1ml/min。
27.进一步地,所述去除单萜类成分的艾草重油成分的检测方法,所述气相色谱-质谱联用仪的质谱条件如下:
28.离子源温度:ei源230℃;
29.扫描质量数范围:m/z:30-500。
30.本发明中检测方法中气相色谱条件与质谱条件相互关联,检测过程是依靠色谱柱固定相与化合物键合后,在本发明色谱条件下,尤其是在上述升温程序下,艾草除萜精油中的大量母核结构类似、沸点相近的组分既能较快的随载气逐一流出色谱柱,又具有一定的分离度;在质谱电压的轰击下,各流出化合物形成离子碎片,进而根据各离子碎片进行定性、定量分析,提高方法的准确度。
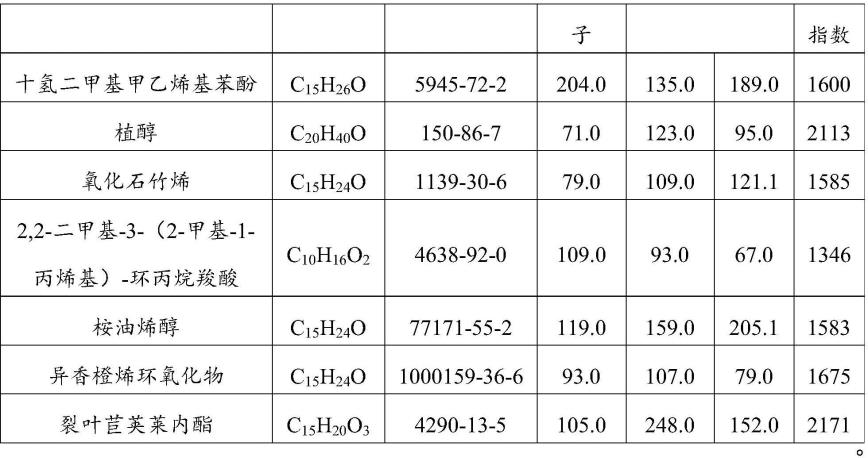
31.进一步地,所述步骤s2在sim模式下对待测物标准品、待测样品成分进行分段扫描得到定量离子和定性离子,扫描结果筛选主要成分、化学式、cas号、定量离子和定性离子(定量离子和定性离子:是化合物进行精确定量时,仪器主要捕捉的质量数,需要成对出现,不同化合物的定量、定性离子对是不同的。同一化合物由于存在的植物来源不同,样品中其他组成物质的干扰也不同,离子化率不同,所选择的离子对也不同)如下表:
32.表1
33.[0034][0035]
超临界分子蒸馏工艺中沸点较高、分子自由程较长的这一段重油中部分成分(如十氢二甲基甲乙烯基苯酚)的检测方法是采用gc或者gcms定性分析的方法进行测定,其无法进行准确定量;而采用本发明提供的定量离子和定性离子捕获的选择离子检测模式则可以精准定量,同时用全扫描模式可以对其他未知化合物进行定性分析。
[0036]
进一步地,所述待测物标准品溶液的制备方法如下:
[0037]
取待测物标准品,用甲醇溶解制成浓度范围为10ppb-1ppm的定量分析标准系列上机溶液;
[0038]
所述c9-c40正构烷烃标准品溶液的制备方法如下:
[0039]
取c9-c40正构烷烃标准品,用正己烷溶解至浓度为100ppb-1ppm,作为定性分析标准溶液;
[0040]
所述待测样品溶液的制备方法如下:
[0041]
待测样品为去除单萜类成分的艾草重油,将待测样品加热至40-90℃,取待测样品适量、转移至容量瓶并用甲醇稀释后,于0~10℃静置15-30min,随后取出用甲醇定容至刻度,制成浓度为0.5~10mg/ml溶液;而后使用0.22μm~0.45μm微孔滤膜过滤即得待测样品溶液。
[0042]
更进一步地,所述待测物标准品溶液的制备方法如下:
[0043]
取待测物标准品,用甲醇溶解制成浓度范围为10ppb、20ppb、40ppb、80ppb、100ppb、200ppb、400ppb的定量分析标准系列上机溶液;
[0044]
所述c9-c40正构烷烃标准品溶液的制备方法如下:
[0045]
取c9-c40正构烷烃标准品,用正己烷溶解至浓度为100ppb作为定性分析标准溶液;
[0046]
所述待测样品溶液的制备方法如下:
[0047]
将待测样品加热至50℃,取待测样品称量、转移至容量瓶并用甲醇稀释后,于4℃静置20min,随后取出用甲醇定容至刻度,制成浓度为0.5~10mg/ml溶液,使用0.22μm微孔
滤膜过滤即得待测样品溶液。
[0048]
此外,本发明还提供一种无标准品半定量分析方法,在缺少某类化合物标准品时可通过半定量分析,进行虚拟定量能够对比批次间样品含量差异。
[0049]
该无标准品半定量分析方法根据检索结果(使用仪器定性分析软件化合物分析的检索功能,获得的检索结果),采集捕获上述无标准品化合物植醇、氧化石竹烯、桉油烯醇、2,2-二甲基-3-(2-甲基-1-丙烯基)-环丙烷羧酸、异香橙烯环氧化物、香附内酯等的定量离子和定性离子,根据样品中无标准品化合物的保留时间和质谱图,建立虚拟标准样品定量离子、定性离子响应曲线,根据待测样品中无标准品化合物的响应,计算模拟浓度,可用于批次间差异对比。
[0050]
该采用无标准品半定量分析方法,增加全扫描scan扫描质量数范围:30-300m/z。利用gcms的nist检索谱图中的各组分的化学结构,通过检索结果和各流出组分的质谱图,采用定量分析同样的思路,选择参照重油中非精确定量组分的保留时间,建立非精确定量组分的采集时间、定量离子和定性离子,设定各组分的浓度为1,即可模拟计算各批次重油中非精确定量组分的模拟浓度。该采用半定量分析方法能够减少仪器污染且可减少多种化合物标准溶液的购买及多种化合物标准溶液逐一配制繁复,具有较高的准确定和精密度,且稳定性及重复性好。
[0051]
进一步地,所述待测物标准品为十氢二甲基甲乙烯基苯酚时,其标准品溶液的制备方法如下:
[0052]
步骤a1:取干燥的艾叶粉碎后与95%乙醇混合,料液比为:8:1~15:1,冷浸提取3次,单次浸提时间为20-25h,过滤将提取液浓缩,制成粗浸提物,加少量水至粗浸提物悬浮于水中,使用石油醚进行萃取并浓缩石油醚层萃取液,得浓缩液;
[0053]
步骤a2:将浓缩液与0.8~1.5倍重量的mci硅胶混合,制成小孔树脂mci柱层析,使用体积比为75:25的甲醇-水体系洗脱3个柱体积,弃去洗脱液i;使用体积比为85:25的甲醇-水体系洗脱3个柱体积,收集洗脱液ii并浓缩为流浸膏;
[0054]
步骤a3:将流浸膏进一步经sephadex lc-20凝胶柱层析,甲醇洗脱,洗脱3个柱体积,每0.5个柱体积为一个组分段,以石油醚:乙酸乙酯=8:1为展开剂,以10%-硫酸乙醇溶液为显色剂,使用薄层色谱法将各组分段中出现紫色斑点段流出液合并,并浓缩为浸膏e1;
[0055]
步骤a4:将浸膏e1使用少量60%甲醇溶解制成悬浮液,进一步经ods反相硅胶柱层析,以体积比为55:45的甲醇-水混合溶液洗脱3-6个柱体积,弃去洗脱液iii;使用体积比为60:40的甲醇-水混合溶液洗脱3个柱体积,收集洗脱液iv合并,并浓缩为浸膏e1.1;浸膏e1.1;
[0056]
步骤a5:浸膏e1.1用少量石油醚制成悬浮液,进一步经硅胶柱层析,以体积比为98:2的石油醚-乙酸乙酯混合溶液洗脱3个柱体积,弃去洗脱液v;使用体积比为95:5的石油醚-乙酸乙酯混合溶液洗脱3个柱体积,收集洗脱液vi浓缩制成流浸膏e1.1.1;
[0057]
步骤a6:流浸膏e1.1.1用少量石油醚制成悬浮液,进一步经硅胶柱层析,以体积比为99:1的石油醚-乙酸乙酯混合溶液洗脱3-6个柱体积,弃去洗脱液vii;使用体积比为98:2的石油醚-乙酸乙酯混合溶液洗脱3个柱体积,收集洗脱液viii浓缩即得十氢二甲基甲乙烯基苯酚标准品。
[0058]
进一步地,所述待测物标准品为裂叶苣荚莱内酯时,其标准品溶液的制备方法如
下:
[0059]
步骤b1:取干燥的艾叶粉碎后与95%乙醇混合,料液比为:8:1~15:1,冷浸提取3次,单次浸提时间为20-25h,过滤将浸提液浓缩,制成粗浸提物,加少量水至粗浸提物悬浮于水中,使用乙酸乙酯进行萃取并浓缩乙酸乙酯层萃取液,得浓缩液;
[0060]
步骤b2:将浓缩液经硅胶柱层析,使用体积比为4:1的石油醚-乙酸乙酯混合溶液洗脱3个柱体积,弃去洗脱液i;使用体积比为3:1的石油醚-乙酸乙酯混合溶液洗脱3个柱体积,收集洗脱液ii并浓缩为浸膏e2;
[0061]
步骤b3:浸膏e2用少量甲醇溶解,进一步经mci柱层析,采用体积比为60:40的甲醇-水混合溶液洗脱3个柱体积,收集洗脱液iii并浓缩为浸膏e2.1;
[0062]
步骤b4:浸膏e2.1用少量甲醇溶解,进一步经ods柱层析,以体积比为45:55的甲醇-水洗脱3个柱体积,弃去洗脱液iv;使用体积比为50:50的甲醇-水混合溶液洗脱3个柱体积,收集洗脱液v并浓缩为浸膏e2.1.1;
[0063]
步骤b5:浸膏e2.1.1使用少量甲醇完全溶解,经制备液相色谱仪反相c18色谱柱,以30%乙腈洗脱,收集响应面积最高的峰,浓缩收集液即得裂叶苣荚莱内酯标准品。
[0064]
与现有技术相比,本发明提供的去除单萜类成分的艾草油质量评价的检测方法,具有以下优势:
[0065]
(1)本发明所述的gcms方法能够对去除单萜类成分的艾草重油中的主要成分进行含量测定,具有较高的准确度和精密度,且稳定性和重复性好,能准确、快速地进行定量分析,方法可行性,结果可靠。
[0066]
(2)本发明中检测方法中气相色谱条件与质谱条件相互关联,检测过程是依靠色谱柱固定相与化合物键合后,在本发明色谱条件下,尤其是在上述升温程序下,艾草除萜精油中的大量母核结构类似、沸点相近的组分既能较快的随载气逐一流出色谱柱,又具有一定的分离度;在质谱电压的轰击下,各流出化合物形成离子碎片,进而根据各离子碎片进行定性、定量分析,最终使得检测方法的准确度得以提高。
附图说明
[0067]
图1为实施例1十氢二甲基甲乙烯基苯酚标准品溶液的tlc图;
[0068]
图2为实施例1裂叶苣荚莱内酯标准品溶液的tlc图;
[0069]
图3为实施例2定性分析艾草重油总离子流图;
[0070]
图4为实施例2定性分析c9-c40正构烷烃总离子流图;
[0071]
图5为实施例2定量分析艾草重油中十氢二甲基甲乙烯基苯酚标准溶液定量离子eic图;
[0072]
图6为实施例2定量分析艾草重油中裂叶苣荚莱内酯标准溶液定量离子eic图;
[0073]
图7为实施例2半定量分析艾草重油中11,11-二甲基-4,8-二甲基二环[7.2.0]十一烷-3-醇全扫描23.3212-23.4959min质谱图;
[0074]
图8为实施例2半定量分析艾草重油溶液中11,11-二甲基-4,8-二甲基二环[7.2.0]十一烷-3-醇的eic图;
[0075]
图9为实施例2半定量分析11,11-二甲基-4,8-二甲基二环[7.2.0]十一烷-3-醇的虚拟标准曲线;
[0076]
图10为试验例一:专属性测试标准空白上机溶液、待测样品溶液和十氢二甲基甲乙烯基苯酚标准溶液全扫描scan模式下获得的tic图;
[0077]
图11为试验例一:线性测试十氢二甲基甲乙烯基苯酚标准系列上机溶液及标准空白上机溶液的标准曲线图。
具体实施方式
[0078]
以下通过具体实施方式的描述对本发明作进一步说明,但这并非是对本发明的限制,本领域技术人员根据本发明的基本思想,可以做出各种修改或改进,但是只要不脱离本发明的基本思想,均在本发明的保护范围之内。
[0079]
其中,本发明所用试剂均为常用试剂,均可在常规试剂生产销售公司购买。
[0080]
实施例1待测物标准品的制备
[0081]
1)标准品溶液的制备:
[0082]
①
十氢二甲基甲乙烯基苯酚标准品溶液的制备方法:
[0083]
步骤a1:取干燥的艾叶粉碎后与95%乙醇混合,料液比为10:1,冷浸提取3次,单次浸提时间为24h,过滤将提取液浓缩,制成粗浸提物,加水至粗浸提物悬浮于水中,使用石油醚进行萃取并浓缩石油醚层萃取液,得浓缩液;
[0084]
步骤a2:将浓缩液与1.2倍重量的mci硅胶混合,制成小孔树脂mci柱层析,使用体积比为75:25的甲醇-水体系洗脱3个柱体积,弃去洗脱液i;使用体积比为85:25的甲醇-水体系洗脱3个柱体积,收集洗脱液ii并浓缩为流浸膏;
[0085]
步骤a3:将流浸膏进一步经sephadex lc-20凝胶柱层析,甲醇洗脱,洗脱3个柱体积,每0.5个柱体积为一个组分段,以石油醚:乙酸乙酯=8:1为展开剂,以10%-硫酸乙醇溶液为显色剂,使用薄层色谱法将各组分段中出现紫色斑点段流出液合并,并浓缩为浸膏e1;
[0086]
步骤a4:将浸膏e1使用60%甲醇溶解制成悬浮液,进一步经ods反相硅胶柱层析,以体积比为55:45的甲醇-水混合溶液洗脱5个柱体积,弃去洗脱液iii;使用体积比为60:40的甲醇-水混合溶液洗脱3个柱体积,收集洗脱液iv合并,并浓缩为浸膏e1.1;
[0087]
步骤a5:浸膏e1.1用石油醚制成悬浮液,进一步经硅胶柱层析,以体积比为98:2的石油醚-乙酸乙酯混合溶液洗脱3个柱体积,弃去洗脱液v;使用体积比为95:5的石油醚-乙酸乙酯混合溶液洗脱3个柱体积,收集洗脱液vi浓缩制成流浸膏e1.1.1;
[0088]
步骤a6:流浸膏e1.1.1用石油醚制成悬浮液,进一步经硅胶柱层析,以体积比为99:1的石油醚-乙酸乙酯混合溶液洗脱5个柱体积,弃去洗脱液vii;使用体积比为98:2的石油醚-乙酸乙酯混合溶液洗脱3个柱体积,收集洗脱液viii浓缩即得十氢二甲基甲乙烯基苯酚标准品。
[0089]
②
裂叶苣荚莱内酯标准品溶液的制备方法:
[0090]
步骤b1:取干燥的艾叶粉碎后与95%乙醇混合,料液比为12:1,冷浸提取3次,单次浸提时间为23h,过滤将浸提液浓缩,制成粗浸提物,加水至粗浸提物悬浮于水中,使用乙酸乙酯进行萃取并浓缩乙酸乙酯层萃取液,得浓缩液;
[0091]
步骤b2:将浓缩液经硅胶柱层析,使用体积比为4:1的石油醚-乙酸乙酯混合溶液洗脱3个柱体积,弃去洗脱液i;使用体积比为3:1的石油醚-乙酸乙酯混合溶液洗脱3个柱体积,收集洗脱液ii并浓缩为浸膏e2;
[0092]
步骤b3:浸膏e2用甲醇溶解,进一步经mci柱层析,采用体积比为60:40的甲醇-水混合溶液洗脱3个柱体积,收集洗脱液iii并浓缩为浸膏e2.1;
[0093]
步骤b4:浸膏e2.1用甲醇溶解,进一步经ods柱层析,以体积比为45:55的甲醇-水洗脱3个柱体积,弃去洗脱液iv;使用体积比为50:50的甲醇-水混合溶液洗脱3个柱体积,收集洗脱液v并浓缩为浸膏e2.1.1;
[0094]
步骤b5:浸膏e2.1.1使用甲醇完全溶解,经制备液相色谱仪反相c18色谱柱,以30%乙腈洗脱,收集响应面积最高的峰,浓缩收集液即得裂叶苣荚莱内酯标准品。
[0095]
2)薄层鉴别:
[0096]
分别取上述待测物十氢二甲基甲乙烯基苯酚、裂叶苣荚莱内酯标准品,用甲醇溶解制成浓度为:10mg/ml溶液,即得十氢二甲基甲乙烯基苯酚、裂叶苣荚莱内酯标准品溶液。
[0097]
实验过程:分别吸取十氢二甲基甲乙烯基苯酚、裂叶苣荚莱内酯标准品溶液,使用微量毛细管手工点样于硅胶g薄层板,以石油醚:乙酸乙酯为展开剂,预饱和20min,上行展开,取出晒干,浸入1%香草醛硫酸乙醇迅速取出,90-105℃加热至半点清晰。
[0098]
3)实验结果:
[0099]
十氢二甲基甲乙烯基苯酚标准品溶液以石油醚:乙酸乙酯=8:1为展开剂,预饱和20min,上行展开,取出晒干,浸入1%香草醛硫酸乙醇迅速取出,105℃加热出现紫色斑点,结果如附图1所示,斑点清晰。
[0100]
裂叶苣荚莱内酯标准品溶液以石油醚:乙酸乙酯=2:1为展开剂,预饱和20min,上行展开,取出晒干,浸入1%香草醛硫酸乙醇迅速取出,100℃加热出现深墨绿色斑点,结果如附图2所示,斑点清晰。
[0101]
在实验过程中综合考虑时间、成本等因素后,先对去除单萜类成分的艾草重油成分薄层色谱法定性鉴别,再展开gcms分析。
[0102]
所述薄层色谱法为:精密称取1g去除单萜类成分的艾草重油样品,置于100ml容量瓶中,甲醇溶解并定容至刻度,摇匀。于4℃静置20min,振荡、离心取上清,制成10mg/ml供试品溶液;精密称取上述二甲基甲乙烯基苯酚标准品、裂叶苣荚莱内酯标准品,制成浓度为1mg/ml标准溶液。以石油醚:乙酸乙酯=2.5:1为展开剂,预饱和20min,上行展开,取出晒干,浸入1%香草醛硫酸乙醇迅速取出,100℃加热,艾草重油供试品色谱带中与对照色谱(十氢二甲基甲乙烯基苯酚标准品溶液、裂叶苣荚莱内酯标准品溶液)相应位置处有紫色斑点。
[0103]
所述去除单萜类成分的艾草重油样品的制备:艾草经超临界二氧化碳萃取工艺制备富含单萜类、倍半萜类、脂肪酸类、三萜类、蜡质类水不溶性浸膏,再经过分子蒸馏工艺,根据工艺特点,不挥发性物质自然冷凝排出,可挥发性物质根据分子自由程差异,(如单萜及少量沸点较低的倍半萜烯)需经冷凝后作为轻组分流出,而倍半萜醇、倍半萜烷、长链烃类、脂肪酸类则作为重组分物质流出,所述重组分即为本发明实施例所述的去除单萜类成分的艾草重油。
[0104]
实施例2一种去除单萜类成分的艾草重油成分的检测方法
[0105]
s1:准备:
[0106]
所述待测物标准品溶液的制备方法如下:
[0107]
取待测物标准品,用甲醇溶解制成浓度范围为10ppb、20ppb、40ppb、80ppb、
100ppb、200ppb、400ppb的定量分析标准系列上机溶液;
[0108]
所述c9-c40正构烷烃标准品溶液的制备方法如下:
[0109]
取c9-c40正构烷烃标准品,用正己烷溶解至浓度为100ppb作为定性分析标准溶液;
[0110]
所述待测样品溶液的制备方法如下:
[0111]
将待测样品置于50℃水浴至液体混合状态,精密称取去除单萜类成分的艾草重油1g,置于100ml容量瓶中,甲醇稀释,于4℃静置20min,取出待溶液恢复室温,甲醇定容至刻度,使用0.22μm微孔滤膜过滤即得待测样品溶液。
[0112]
s2:分别将待测物标准品溶液和待测样品溶液以相同的采集方法分别注入气相色谱-质谱联用仪中,同时进行scan全扫描+sim选择离子检测,得到待测物标准品溶液和待测样品溶液中化合物的定量离子及定性离子、离子流图eic以及总离子流图tic;将c9-c40正构烷烃标准品溶液以相同的采集方法注入气相色谱-质谱联用仪中,得c9-c40正构烷烃标准品溶液的总离子流图tic;
[0113]
所述步骤s2中气相色谱-质谱联用仪的色谱条件如下:
[0114]
色谱柱:5%苯基甲基聚硅氧烷为固定相的色谱柱,柱长30m;
[0115]
升温程序:70℃保持2min,以5℃/min升至150℃保持2min,以3℃/min升至250℃保持2min,以10℃/min升至290℃保持1min;
[0116]
进样口温度:240℃;
[0117]
分流比:50:1;
[0118]
所述气相色谱的载气为氦气,流速为1ml/min。
[0119]
所述步骤s2中气相色谱-质谱联用仪的质谱条件如下:
[0120]
离子源温度:ei源230℃;
[0121]
扫描质量数范围:m/z:30-500。
[0122]
所述步骤s2在sim模式下对待测物标准品、待测样品成分进行分段扫描得到定量离子和定性离子,扫描结果筛选主要成分、化学式、cas号、定量离子和定性离子如下表:
[0123]
表2
[0124][0125]
s3:定性分析:将c9-c40正构烷烃标准品溶液的总离子流图tic导入定性分析软件中,使用nist库对数据进行匹配,识别并获得c9-c40正构烷烃的保留时间;将待测样品采集的总离子流图导入分析软件中,采用解卷积计算方法,筛选与谱库匹配度>70%,获得定性分析检索化合物,根据各化合物的保留时间与正构烷烃的保留时间比值计算化合物的保留指数,对比化合物的标准保留指数与谱库检索的匹配度,获得艾草重油定性分析数据结果;其中c9-c40正构烷烃保留指数如下表(下表中仅列出检测条件下能在色谱图上显示的烷烃):
[0126]
表3
[0127]
保留时间化合物名称分子式cas号保留指数4.0907正壬烷c9h
20
111-84-29006.0748正癸烷c
10h22
124-18-510008.535正十一烷c
11h24
1120-21-4110011.1893十二烷c
12h26
112-40-3120013.8612十三烷c
13h28
629-50-5130016.4449十四烷c
14h30
629-59-4140018.9757十五烷c
15h32
629-62-9150022.0746十六烷c
16h34
544-76-3160025.3367十七烷c
17h36
629-78-7170028.6148十八烷c
18h38
593-45-3180031.8486十九烷c
19h40
629-92-5190034.9853二十烷c
20h42
112-95-8200038.0178二十一烷c
21h44
629-94-72100
40.9365二十二烷c
22h46
629-97-0220043.7388二十三烷c
23h48
638-67-5230046.4442二十四烷c
24h50
646-31-1240049.0429二十五烷c
25h52
629-99-2250051.5446二十六烷c
26h54
630-01-3260052.1167二十七烷c
27h56
593-49-7270053.9591二十八烷c
28h58
630-02-4280056.296二十九烷c
29h60
630-03-5290057.3917三十烷c
30h62
638-68-6300058.7298三十一烷c
31h64
630-04-6310060.7758三十二烷c
32h66
544-85-4320061.1928三十三烷c
33h68
630-05-7330062.2594三十四烷c
34h70
14167-59-03400
[0128]
;
[0129]
去除单萜类成分的艾草重油定性结果如下表(下表中化合物名称采用仪器直接导出的名称,命名方式符合国际命名原则):
[0130]
表4
[0131]
[0132]
[0133]
[0134]
[0135]
[0136][0137]
s4:定量分析:根据sim选择离子检测数据结果,使用定量分析软件,对质谱图进行分析,并分别对选择的定量离子、定性离子进行积分,获取选定待测物标准品溶液与待测样品响应比值、标准品溶液浓度,计算出待测物的浓度;使用定量分析软件,对质谱图进行自动分析。
[0138]
s5:半定量分析:在进行定性分析以后,根据检索结果,发现结果中除精确定量的主要成分(2,2-二甲基-3-(2-甲基-1-丙烯基)-环丙烷羧酸、桉油烯醇、氧化石竹烯、十氢二甲基甲乙烯基苯酚、异香橙烯环氧化物、植醇、裂叶苣荚莱内酯)以外,保留时间为23.3838min的定性分析成分11,11-二甲基-4,8-二甲基二环[7.2.0]十一烷-3-醇(11,11-dimethyl-4,8-dimethylenebicyclo[7.2.0]undecan
[0139]-3-ol)的峰面积相对于其他成分较大,预测其可能含量较高,但因无标准品,又需要对多个批次中的该成分进行定量预估,以判断各批次间含量变化差异,因此进行半定量分析。
[0140]
根据待测样品溶液的全扫描定性分析总离子流图,捕获保留时间为23.3212-23.4959min的质谱图,见图7。
[0141]
根据其质谱图,筛选模拟定量离子及定性离子:136.0m/z、187.0m/z及105.0m/z,进入定量分析软件,根据之前定量分析结果,以浓度为100ppb的十氢二甲基甲乙烯基苯酚标准溶液的响应值为模拟值,建立强制过原点的单点虚拟校正曲线,设定该虚拟响应值下11,11-二甲基-4,8-二甲基二环[7.2.0]十一烷-3-醇的浓度为1。即可获得模拟定量待测样品溶液中11,11-二甲基-4,8-二甲基二环[7.2.0]十一烷-3-醇化合物浓度相对于十氢二甲基甲乙烯基苯酚的浓度,从而对比批次间艾草重油该化合物浓度。下表为11,11-二甲基-4,8-二甲基二环[7.2.0]十一烷-3-醇的虚拟定量结果。
[0142]
表5
[0143]
批号保留时间响应值虚拟浓度1030-0123.3831556080.7319
1027-0223.3841697000.79821027-0123.3841826580.85921022-0123.3831400520.65881018-0123.3831255810.59071011-0223.3841198610.56381011-0123.3841463930.6886
[0144]
实验结果:
[0145]
如图3所示为实施例2定性分析步骤s3所得的艾草重油总离子流图。
[0146]
如图5所示为实施例2定性分析艾草重油中十氢二甲基甲乙烯基苯酚标准溶液定量离子eic图。其中,左图十氢二甲基甲乙烯基苯的实际保留时间为24.025min时其离子流出图中定量离子204.1m/z;而右图定性离子135.1m/z和189.1m/z在24.025min时与定量离子204.1m/z同时流出,是精确定量,定量结果无干扰的表现之一。
[0147]
如图6所示为实施例2定性分析艾草重油中裂叶苣荚莱内酯标准溶液定量离子eic图。其中,左图裂叶苣荚莱内酯的实际保留时间为40.108min时其离子流出图中定量离子248.1m/z;而右图定性离子152.1m/z和107.1m/z在40.108min时与定量离子248.1m/z同时流出,同样是精确定量,定量结果无干扰的表现之一。
[0148]
如图7所示为实施例2半定量分析艾草重油中11,11-二甲基-4,8-二甲基二环[7.2.0]十一烷-3-醇全扫描23.3212-23.4959min质谱图。
[0149]
如图8所示为实施例2半定量分析艾草重油溶液中11,11-二甲基-4,8-二甲基二环[7.2.0]十一烷-3-醇的eic图。
[0150]
试验例一:性能测试
[0151]
以十氢二甲基甲乙烯基苯酚为例展开本发明去除单萜类成分的艾草重油质量评价检测方法的专属性、适应性、准确度、精密度等性能测试。
[0152]
(1)专属性
[0153]
分别准备标准空白上机溶液即试剂空白溶液、十氢二甲基甲乙烯基苯酚标准溶液和待测样品溶液,按相同测试条件进样分析。
[0154]
结果:如图10所示,tic scan blk-002为标准空白上机溶液组,tic scan 1009-009为待测样品溶液组,tic scan 1009-008为十氢二甲基甲乙烯基苯酚标准溶液组。标准空白上机溶液对样品无定量干扰,测定方法对样品测定无干扰,表明本发明所提供的去除单萜类成分的艾草重油质量评价检测方法的专属性极强。
[0155]
(2)适应性
[0156]
取十氢二甲基甲乙烯基苯酚标准系列上机溶液为质量检验标准(qc check standard),每进样不超过10次及进样结束均测一次质量检验标准,比较待测样品溶液测试前后所得质量检验标准,计算飘移值。
[0157]
qc飘移值=8<10%。
[0158]
(3)线性
[0159]
取十氢二甲基甲乙烯基苯酚标准系列上机溶液及标准空白上机溶液进样分析,计算获得校准曲线和相关系数。
[0160]
结果:如图11所示,得线性方程y=50389.837231*x-144333.822970,r2=0.9986,
其中相关系数>0.99,相关性较强。
[0161]
(4)准确度
[0162]
将待测样品去除单萜类成分的艾草重油置于50℃水浴至液体混合状态,随后精密称取去除单萜类成分的艾草重油1g,置于100ml容量瓶中,加入十氢二甲基甲乙烯基苯酚标准品适量,甲醇稀释,于4℃静置20min,取出待溶液恢复室温,甲醇定容至刻度,使用0.22μm微孔滤膜过滤制成加标浓度为50%、100%、150%的低、中、高浓度加标溶液,各浓度梯度样品≥3份,逐一进样分析,计算样品中组分的低、中、高浓度加标回收率。低、中、高浓度加标回收率平均值分别为:104.6%、98.2%、102.1%。
[0163]
回收率均在80%-120%之间。
[0164]
(5)精密度
[0165]
取平行制备的6份相同浓度去除单萜类成分的艾草重油样品溶液进样分析,计算样品中待测成分测得量的相对标准偏差rsd%(n=6)。rsd=1.6%<2%,精密度较高。
[0166]
上述实施例仅例示性说明本发明的原理及其功效,而非用于限制本发明。任何熟悉此技术的人士皆可在不违背本发明的精神及范畴下,对上述实施例进行修饰或改变。因此,举凡所属技术领域中具有通常知识者在未脱离本发明所揭示的精神与技术思想下所完成的一切等效修饰或改变,仍应由本发明的权利要求所涵盖。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1