一种甲氨蝶呤血药浓度的检测方法
1.本发明属于药物分析检测技术领域,具体涉及一种甲氨蝶呤血药浓度的检测方法。
背景技术:
2.甲氨蝶呤(methotrexate,mtx),又名氨甲喋呤,是一种二氢叶酸还原酶抑制剂,作用于g1及g1/s转换细胞,阻止dna及rna的合成,其作为临床联合化疗方案中常用的细胞周期特异性抗代谢类药物,广泛应用于儿童急性淋巴细胞白血病、淋巴瘤、绒毛膜上皮癌和骨肉瘤等恶性肿瘤的治疗。由于mtx对肿瘤细胞的作用缺乏选择性,在杀灭肿瘤细胞的同时会杀灭正常细胞,随着临床应用的逐渐增多,mtx的骨髓抑制、神经毒性(鞘内给药)、肾小管阻塞和损伤等副作用也逐渐引起临床医生的重视。产生这些副作用的直接原因是化疗时患者体内的mtx血药浓度过高且持续时间过长,这与mtx的大剂量给药方式有关。幸运的是,在应用mtx后,可通过外源给与亚叶酸钙,越过mtx阻断的部位使dna和蛋白质的合成正常进行,从而起到解救作用。因此,及时检测mtx血药浓度并根据其浓度及时调整亚叶酸钙解救的时间和剂量,可以有效减少副作用。
3.目前,mtx血药浓度的检测方法主要有紫外光谱法、毛细管电泳法、色谱法、质谱法、均相酶免疫法和荧光偏振免疫分析法。虽然这些方法均可用于mtx血药浓度的测定,但在检测中也有一定的局限性:紫外光谱法操作简单但易受血中蛋白及其他共存物质的干扰,选择性欠佳;毛细管电泳法所需样本量少但同样易受其他共存物质的影响,因此在选择性上需要进一步改进;色谱法是金标方法,但前处理需经固相萃取柱处理,过程比较繁琐;质谱法具有较好的分离及定性定量特性,但仪器相对昂贵,且需要复杂的样本预处理过程,仪器不易得且分析速度较慢;均相酶免疫分析法的仪器要求简单但需要较长时间的提取过程,提取程度对实验结果影响较大,不同操作人员检测结果的一致性较差,分析速度和精密度均有限;荧光偏振免疫分析法操作简便快速,但交叉反应对其影响较大,可见方法灵敏度与选择性仍需进一步提高。
技术实现要素:
4.本发明的目的在于克服现有技术的不足,提供一种甲氨蝶呤血药浓度的检测方法。
5.为实现上述目的,本发明采用的技术方案是:
6.一种甲氨蝶呤血药浓度的检测方法,包括以下步骤:
7.s1、将甲醇加入到不含甲氨蝶呤(mtx)的正常血清中,超声混匀并离心,留取上清液,得到a液;
8.s2、将50~150mg的磁性分子印迹聚合物(mmips)加入甲醇中振荡活化,经磁性分离,过滤掉上清液,得到反应物;
9.s3、将步骤s1中得到的a液和步骤s2中得到的反应物分别加入到多组不同浓度的
甲氨蝶呤标准溶液中,得到多组混合物,分别于室温下振荡120~480min萃取上样,磁性分离,回收上样,接着加入淋洗液振荡除杂,得到多组处理后的材料;
10.s4、将3~6ml的洗脱液分别加入到步骤s3中得到的多组材料中,振荡40~120min,经磁性分离后,留取上清液,利用n2吹干并加入500μl甲醇复溶,然后过滤,将多组滤液分别通过hplc-uv法进行检测,得到标准曲线;
11.s5、将步骤s3中的不同浓度的甲氨蝶呤标准溶液更换为实际血样样品,接着重复步骤s3和步骤s4,将步骤s4中得到的滤液通过hplc-uv法进行检测,然后根据步骤s4得到的标注曲线计算得到实际血样样品中的甲氨蝶呤血药浓度。
12.优选的,步骤s3中,所述甲氨蝶呤标准溶液的浓度为0.00005~0.25mg/ml。
13.优选的,步骤s3中,多组所述混合物中的甲氨蝶呤的浓度均为0.000004~0.02mg/ml。
14.优选的,步骤s3中,所述淋洗液为水-甲醇,且水和甲醇的体积比为4:1。
15.优选的,步骤s4中,所述洗脱液为甲醇-乙酸,且甲醇和乙酸的体积比为4:1。
16.优选的,步骤s4中,所述过滤步骤采用0.22μm的有机滤膜过滤。
17.优选的,步骤s4中,所述hplc-uv法的检测条件为:检测波长306nm,流速1ml/min,进样量20μl,温度30℃。
18.本发明与现有技术相比,其有益效果在于:
19.(1)本发明提供的检测方法由于磁性纳米颗粒的引入,固相萃取分离的过程采用外加磁场下的磁性分离,1分钟左右即可将萃取溶液与固相萃取剂分离,代替了常规发光检测对样本的离心处理,节省了仪器成本与分离时间;与常规固相萃取方法相比,由于磁性纳米颗粒的引入,磁性固相萃取不需要常规的固相萃取仪,节省了仪器成本;
20.(2)本发明中的磁性固相萃取用的固相萃取材料为自制的磁性分子印迹聚合物,能够将目标分子甲氨蝶呤特异性的吸附于形成的印迹孔穴中,相比于常规固相萃取剂对甲氨蝶呤的吸附具有更好的选择性;
21.(3)本发明采用磁性固相萃取-液相色谱联用的方法,用于实际血浆样品中mtx的检测,具有准确性和实用性,将其检测的实际样本中甲氨蝶呤浓度结果与临床所用其他方法进行比较,具有较好的一致性,同时本发明提供的检测方法可以克服均相酶免疫方法中交叉反应的局限性,结果更加准确。
附图说明
22.图1为本发明实施例1-5以及对比例1-4提供的检测方法的萃取效果图;
23.图2为本发明实施例1、6和7与对比例5-8提供的检测方法的萃取效果图;
24.图3为不同乙酸含量的洗脱液下的mtx萃取回收率图;
25.图4为不同洗脱液体积下的mtx萃取回收率图;
26.图5为不同洗脱时间下的mtx萃取回收率图;
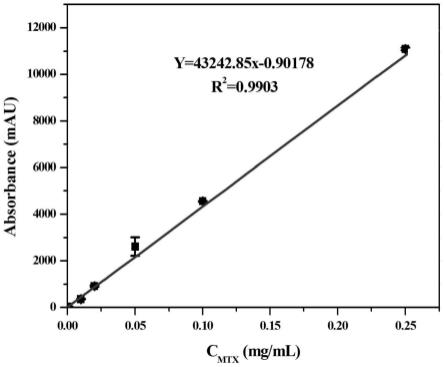
27.图6为本发明实施例提供的检测方法中的标准曲线图;
28.图7为相同浓度下mtx标准溶液与经mmips和mnips萃取后的加标溶液的色谱图;
29.其中:图7(a)为mtx标准溶液(0.05mg/ml)萃取后的色谱图;图7(b)为mmips萃取后的加标溶液(0.05mg/ml)的色谱图;图7(c)为mnips萃取后的加标溶液(0.05mg/ml)的色谱
图。
具体实施方式
30.下面将参照附图更详细地描述本公开的示例性实施例。虽然附图中显示了本公开的示例性实施例,然而应当理解,可以以各种形式实现本公开而不应被这里阐述的实施例所限制。相反,提供这些实施例是为了能够更透彻地理解本公开,并且能够将本公开的范围完整的传达给本领域的技术人员。
31.实施例1
32.本发明实施例提供的甲氨蝶呤血药浓度的检测方法,具体包括以下步骤:
33.s1、将10ml甲醇加入到50μl不含甲氨蝶呤(mtx)的正常血清中,超声混匀后,以8000rpm离心5min,留取上清液,得到a液;
34.s2、将100mg的mmips置于15ml离心管中,加入10ml甲醇中振荡活化5min,活化完全后,经磁性分离,过滤掉上清液,得到反应物,可配置多组反应物;
35.s3、将4.6ml步骤s1中得到的a液和400μl步骤s2中得到的反应物分别加入到400μl的浓度为0.00005、0.010、0.020、0.05、0.10和0.25mg/ml的mtx标准溶液中,得到多组混合物,多组混合物的终浓度分别为0.000004、0.0008、0.0016、0.004、0.008和0.02mg/ml,分别于室温下振荡120min萃取上样,磁性分离,回收上样,接着加入5ml的水-甲醇淋洗液(4:1,v/v)振荡1min,洗去残留的杂质,得到多组处理后的材料;
36.s4、将5ml的甲醇-乙酸(4:1,v/v)洗脱液分别加入到步骤s3中得到的多组材料中,振荡60min,经磁性分离后,留取上清液,将液体倒入试管中,利用n2吹干并加入500μl甲醇复溶,然后采用0.22μm的有机滤膜过滤,将多组滤液分别通过hplc-uv法进行检测,得到标准曲线;
37.s5、将步骤s3中的不同浓度的甲氨蝶呤标准溶液更换为实际血样样品,接着重复步骤s3和步骤s4,将步骤s4中得到的滤液通过hplc-uv法进行检测,然后根据步骤s4得到的标注曲线计算得到实际血样样品中的甲氨蝶呤血药浓度。
38.上述hplc-uv法的检测条件为:检测波长306nm,流速1ml/min,进样量20μl,温度30℃,具体高效液相色谱流动相参数如下表1所示。
39.表1高效液相色谱流动相参数表
[0040][0041]
实施例2
[0042]
本发明实施例提供的检测方法与实施例1的检测方法相同,区别仅在于,步骤s2
中,所述mmips的加入量为50mg。
[0043]
实施例3
[0044]
本发明实施例提供的检测方法与实施例1的检测方法相同,区别仅在于,步骤s2中,所述mmips的加入量为150mg。
[0045]
实施例4
[0046]
本发明实施例提供的检测方法与实施例1的检测方法相同,区别仅在于,步骤s3中,所述振荡时间为240min。
[0047]
实施例5
[0048]
本发明实施例提供的检测方法与实施例1的检测方法相同,区别仅在于,步骤s3中,所述振荡时间为480min。
[0049]
实施例6
[0050]
本发明实施例提供的检测方法与实施例1的检测方法相同,区别仅在于,步骤s3中,所述淋洗液中水和甲醇的体积比为7:3。
[0051]
实施例7
[0052]
本发明实施例提供的检测方法与实施例1的检测方法相同,区别仅在于,步骤s3中,所述淋洗液中水和甲醇的体积比为9:1。
[0053]
实施例8
[0054]
本发明实施例提供的检测方法与实施例1的检测方法相同,区别仅在于,步骤s4中,所述洗脱液中的甲醇和乙酸的体积比为9:1。
[0055]
实施例9
[0056]
本发明实施例提供的检测方法与实施例1的检测方法相同,区别仅在于,步骤s4中,所述洗脱液中的甲醇和乙酸的体积比为7:3。
[0057]
实施例10
[0058]
本发明实施例提供的检测方法与实施例1的检测方法相同,区别仅在于,步骤s4中,所述洗脱液的体积为3ml。
[0059]
实施例11
[0060]
本发明实施例提供的检测方法与实施例1的检测方法相同,区别仅在于,步骤s4中,所述洗脱液的体积为4ml。
[0061]
实施例12
[0062]
本发明实施例提供的检测方法与实施例1的检测方法相同,区别仅在于,步骤s4中,所述洗脱液的体积为6ml。
[0063]
实施例13
[0064]
本发明实施例提供的检测方法与实施例1的检测方法相同,区别仅在于,步骤s4中,所述振荡时间为40min。
[0065]
实施例14
[0066]
本发明实施例提供的检测方法与实施例1的检测方法相同,区别仅在于,步骤s4中,所述振荡时间为120min。
[0067]
对比例1
[0068]
本对比例提供的检测方法与实施例1的检测方法相同,区别仅在于,步骤s2中,所
述mmips的加入量为30mg。
[0069]
对比例2
[0070]
本对比例提供的检测方法与实施例1的检测方法相同,区别仅在于,步骤s2中,所述mmips的加入量为20mg。
[0071]
对比例3
[0072]
本对比例提供的检测方法与实施例1的检测方法相同,区别仅在于,步骤s3中,所述振荡时间为50min。
[0073]
对比例4
[0074]
本对比例提供的检测方法与实施例1的检测方法相同,区别仅在于,步骤s3中,所述振荡时间为25min。
[0075]
对比例5
[0076]
本对比例提供的检测方法与实施例1的检测方法相同,区别仅在于,步骤s3中,所述淋洗液为水-乙腈,且水和乙腈的体积比为9:1。
[0077]
对比例6
[0078]
本对比例提供的检测方法与实施例1的检测方法相同,区别仅在于,步骤s3中,所述淋洗液为水-乙腈,且水和乙腈的体积比为8:2。
[0079]
对比例7
[0080]
本对比例提供的检测方法与实施例1的检测方法相同,区别仅在于,步骤s3中,所述淋洗液为水-乙腈,且水和乙腈的体积比为7:3。
[0081]
对比例8
[0082]
本对比例提供的检测方法与实施例1的检测方法相同,区别仅在于,步骤s3中,所述淋洗液为水。
[0083]
对比例9
[0084]
本对比例提供的检测方法与实施例1的检测方法相同,区别仅在于,步骤s4中,所述洗脱液中的甲醇和乙酸的体积比为19:1。
[0085]
对比例10
[0086]
本对比例提供的检测方法与实施例1的检测方法相同,区别仅在于,步骤s4中,所述洗脱液的体积为2ml。
[0087]
对比例11
[0088]
本对比例提供的检测方法与实施例1的检测方法相同,区别仅在于,步骤s4中,所述振荡时间为20min。
[0089]
对比例12
[0090]
本对比例提供的检测方法与实施例1的检测方法相同,区别仅在于,步骤s4中,所述振荡时间为10min。
[0091]
对比例13
[0092]
本对比例提供的检测方法与实施例1的检测方法相同,区别仅在于,步骤s4中,所述振荡时间为5min。
[0093]
对比例14
[0094]
本对比例提供的检测方法与实施例1的检测方法相同,区别仅在于,步骤s2中,
mmips更换为mnips(没有印迹聚合物的磁性分子)。
[0095]
对比例15
[0096]
本对比例提供的检测方法与实施例2的检测方法相同,区别仅在于,步骤s2中,mmips更换为mnips(没有印迹聚合物的磁性分子)。
[0097]
对比例16
[0098]
本对比例提供的检测方法与实施例3的检测方法相同,区别仅在于,步骤s2中,mmips更换为mnips(没有印迹聚合物的磁性分子)。
[0099]
对比例17
[0100]
本对比例提供的检测方法与对比例1的检测方法相同,区别仅在于,步骤s2中,mmips更换为mnips(没有印迹聚合物的磁性分子)。
[0101]
对比例18
[0102]
本对比例提供的检测方法与对比例2的检测方法相同,区别仅在于,步骤s2中,mmips更换为mnips(没有印迹聚合物的磁性分子)。
[0103]
一、本发明实施例1-14和对比例1-14提供的检测方法的检测效果对比研究
[0104]
1、萃取剂mmips的用量对萃取效果的影响
[0105]
图1(a)为本发明实施例1-3和对比例1-2以及对比例14-18的检测方法的萃取效果图,通过图1(a)可以看出,当mmips用量从20mg增加到100mg时,色谱图峰面积逐渐增加,进一步增加用量到150mg时峰面积无明显增加,而mnips用量在50mg时即可达到最大吸附,且其吸附量远小于mmips,这是因为mmips印迹了模板分子后,在洗掉模板后重新吸附模板分子的时候对模板分子的吸附量会明显高于磁性非印迹聚合物mnips,因此,选择mmips作为吸附剂,为了获得最佳的mtx上样效果,mmips用量选择100mg。
[0106]
2、萃取时间对萃取效果的影响
[0107]
图1(b)为本发明实施例1、4和5与对比例3和4的检测方法的萃取效果图,通过图1(b)可以看出,当萃取时间为120分钟时,萃取剂的吸附效果达到最佳,当萃取时间大于120分钟时,mtx的色谱图峰面积保持稳定,而当萃取时间小于120分钟时,萃取的效果并不是很好,即本发明实施例1、4和5提供的检测方法的萃取效果要优于对比例3和4的萃取效果,因此,本发明选择120分钟作为萃取上样的最佳萃取时间。
[0108]
3、淋洗液的类型对萃取效果的影响
[0109]
淋洗液的选择需要遵循两个原则,一方面应最大程度去除留存于mmips上的干扰物质,另一方面确保淋洗时mtx的损失最少。图2为本发明实施例1、6和7与对比例5-8的检测方法的萃取效果图,通过测量淋洗后上清液中mtx的浓度,来探究不同体积比的淋洗液对mspe效果的影响。通过图2可以看出,当淋洗液为水-甲醇,且水和甲醇的体积比为8:2时,mmips与mnips的色谱峰面积相差最小,即mmips的淋洗液中mtx含量较少,mnips的淋洗液中mtx含量较多,表明此淋洗液的淋洗效果最佳。因此,本发明选择水:甲醇(8:2,v/v)作为淋洗液。
[0110]
4、洗脱液中不同乙酸含量对mtx萃取回收率的影响
[0111]
本发明实施例采用不同体积分数的甲醇-乙酸溶液作为洗脱液,用以提取吸附在mmips和mnips上的mtx。图3为不同乙酸含量的洗脱液下的mtx萃取回收率图,通过图3可以看出,本发明实施例采用甲醇-乙酸溶液作为洗脱液提取吸附在mmips上的mtx量远大于
mnips上的量,且当甲醇-乙酸的体积比为8:2时,萃取回收率最大。
[0112]
5、洗脱液体积对mtx萃取回收率的影响
[0113]
图4为不同洗脱液体积下的mtx萃取回收率图,通过图4的结果可以看出,当洗脱液体积由2ml增加到5ml,回收率由64.65%提高到93.51%,随着洗脱液体积进一步增大,萃取回收率无明显增加。因此,选择5ml作为最佳的洗脱液体积。
[0114]
6、洗脱时间对mtx萃取回收率的影响
[0115]
图5为不同洗脱时间下的mtx萃取回收率图,通过图5的结果可以看出,当洗脱时间少于60min时,萃取回收率较低,当洗脱时间为60min时,萃取回收率趋于稳定,之后不再增加。因此,选择60min作为mspe过程的最佳洗脱时间。
[0116]
二、本发明实施例1提供的检测方法(mspe-hplc-uv)的评价及应用研究
[0117]
本发明绘制了mspe-hplc-uv方法中检测mtx加标溶液的标准曲线,如图6所示。通过图6结果可知,加标法测得mtx在0.00005-0.25mg/ml范围内呈线性,线性方程为y=43242.85c(mg/ml)-0.90178(r2=0.9903)。根据信噪比(s/n=3)得出,该传感器的检测限为12.51ng/ml。该方法检测限低于mtx加标浓度,可以满足mtx血药浓度检测的需要,在一定程度上丰富了mtx的检测方法。
[0118]
下面对本发明实施例1提供的mspe-hplc-uv方法与现有仪器实验结果进行对比研究
[0119]
现有检测mtx血药浓度的常用仪器是药物浓度分析仪(西门子,上海),该检测仪器的基本原理是酶放大免疫测定法(enzyme multiplied immunoassay technique,emit)。通过本发明实施例1提供的mspe-hplc-uv检测方法检测实际血浆样品中mtx的含量,并将检测结果与药物浓度分析仪检测结果进行比对,结果见下表2,对两种方法的检测结果进行wilcoxon分析,结果见下表3。
[0120]
表2两种方法检测mtx血药浓度的结果对比
[0121]
[0122][0123]
表3两种方法检测mtx血药浓度的wilcoxon分析结果
[0124][0125]
通过表3的结果可知,p=0.360》0.05,说明两种方法检测结果差异无统计学意义,即结果一致性较好。因此,本发明实施例1建立的mspe-hplc-uv方法对mtx检测的准确性较好,且与酶放大免疫测定法相比,不存在免疫交叉反应,准确性强,且本发明实施例的检测方法前处理分离方法简单。
[0126]
相同浓度下mtx标准溶液与经mmips和mnips萃取后的加标溶液的色谱图如图7所示,13min处为mtx的特征峰,经mmips萃取的样品色谱图基线较平滑,与同等浓度mtx标准溶液的色谱图峰型相似、峰面积相近,而经mnips萃取后的色谱图峰面积远小于标准溶液。这表明mspe-hplc-uv方法可以抵抗血浆中复杂基质的干扰,且能起到有效富集mtx的作用。进一步证实本发明实施例1建立的检测方法具有准确性和实用性,可用于实际血浆样品中mtx的检测。
[0127]
综上所述,本发明实施例提供的甲氨蝶呤血药浓度的检测方法具有准确性和实用性,可用于实际血浆样品中mtx的检测。
[0128]
尽管已经示出和描述了本发明的实施例,本领域的普通技术人员可以理解:在不脱离本发明的原理和宗旨的情况下可以对这些实施例进行多种变化、修改、替换和变型,本发明的范围由权利要求及其等同物限定。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1