一种亚稳相稀土镍基氧化物氢致电子相变动力学调控方法
1.本发明属于材料科学与电子信息领域,具体地涉及一种通过稀土镍基氧化物与金属间的功函数差异与溶液ph值的综合设计,实现稀土镍基氧化物氢致电子相变程度与速率的动力学调控方法。
背景技术:
2.稀土镍基钙钛矿氧化物是典型的处于热力学亚稳相结构的强关联半导体,其化学式通常用renio3(re=rare earth,re≠la)表示,具有典型的金属绝缘相变输运特性,即当温度升高到某一临界值时,即从绝缘相转变为金属相【nat.commun.,2014,5,4860;appl.phys.lett.,2015,107,031905;phys.rev.lett.,1999,82,3871;phys.rev.b,2004,69,153105;phase transitions,2008,81,729】。这一特点使得该材料在突变式热敏电阻【adv.funct.mater.,2020,30,2000987】、红外伪装【proc.natl.acad.sci,2019,116,26402】以及固体氧化物燃料电池【nature,2016,534,231】等领域内具有广阔的运用前景。
3.除了温致金属绝缘相变特性,稀土镍基钙钛矿氧化物材料的另一特点是具有响应迅速且可逆的氢致相变特性【nat.commun.,2014,5,4860】。其特征在于,在铂催化剂协同下通过氢元素可逆掺杂使得材料由原有的基于+3价镍的电子巡游态轨道构型转变为基于+2价镍的电子局域态电子轨道构型,并触发电阻或电阻率发生突变。上述特性可以通过稀土镍基氧化物在氢气气氛下退火实现,亦可通过稀土镍基氧化物在电化学溶液环境中通过电场触发实现。
4.氢致相变过程主要过程如下:首先,在稀土镍基氧化物薄膜材料上生长一定形状的贵金属如pt;其次,在一定的温度如300℃和低氢气浓度下(如5%)反应一定的时间便可实现电阻率的突变;最后,其逆过程一般是在氧气气氛下进行的。稀土镍基氧化物的氢致电子相变特性在非易失仿生神经网络【nat.commun.,2017,8,240】,突触晶体管【nat.commun.,2013,4,2676】和生物质传感【nat.commun.,2019,10,1651】等领域具有重要的应用价值。此外,对于空穴掺杂的稀土镍基氧化物材料如nd
0.8
sr
0.2
nio3等,将该材料和一定量的cah2固体放置在一个密闭的环境中,在一定温度下反应一定时间,之后进行电输运测试,便可在一定温度下实现超导【nature,2019,572,624】,这一特性也可以看做是一种氢致相变。
5.虽然氢致相变对于稀土镍基氧化物的电输运特性具有重要的调控作用,以及广阔的应用前景,然而当前实现对稀土镍基氧化物的氢致相变方法目前仅有的两种触发方式难以实现对氢致电子相变动力学过程的精准控制。例如,在无论是氢气退火还是电化学溶液中的电场触发所实现的氢致相变,均难以实现对氢致电阻变化速率的准确设计与控制。挖掘能够精准控制亚稳相强关联量子材料体系氢致相变新的新触发方式并实现电子相变动力学的准确控制,将能够进一步拓展该材料在其他领域内的应用,比如人体健康监测、酸碱气体监测、工业废水酸碱性达标监测等。
技术实现要素:
6.本发明主要涉及一种通过亚稳相稀土镍基氧化物与金属间的功函数差异与溶液ph值的综合设计,实现对稀土镍基氧化物氢致电子相变程度与速率的动力学特性的调控方法。
7.本发明通过将金属电极和稀土镍基氧化物相接触并放置在酸性或碱性的化学环境中来调控稀土镍基氧化物氢致电子相变。一方面,利用金属电极和稀土镍基氧化物功函数之间的合理匹配,结合酸性或碱性化学环境程度的选择,实现对稀土镍基氧化物氢致电子相变程度和速率的精准设计,实现其在类脑计算、仿生神经等领域内的应用;另一方面,基于金属电极和稀土镍基氧化物在不同ph值的化学环境中氢致电子相变特性的变化规律,可实现对溶液、气体、雾化气氛等化学环境中酸碱度的探测。
8.一种亚稳相稀土镍基氧化物氢致电子相变动力学调控方法,将特定金属电极以可控形状与稀土镍基氧化物材料相接触,并放置在酸性化学环境中,基于金属和稀土镍基氧化物材料功函数间的差异,驱使化学环境中的氢离子发生转移,从而实现稀土镍基氧化物材料的电子构型以可控的速率由以三价镍为主导的巡游态电子结构向以二价镍为主导的局域态电子结构转变,并触发材料电阻率按可控速率而增加,这种可控性的实现方式包括稀土元素、电极种类、电极形状以及ph值的改变。当特定金属电极以可控形状与稀土镍基氧化物材料相接触,并放置在碱性化学环境中时,将触发材料电阻率的急剧减小。
9.通过施加电场可以进一步精确调控这一氢致电子相变过程;这一新的触发氢致相变的方法可以实现对稀土镍基氧化物氢致电子相变程度和速率的精准设计和对酸性或碱性化学环境的探测。
10.进一步地,所述的稀土镍基钙钛矿氧化物材料的晶体结构为abo3的钙钛矿结构:a位为单一稀土元素或与稀土元素多种稀土元素的组合,优选镝:dy,铒:er,镱:yb,钐:sm,钕:nd,铕:eu,镨:pr,镧:la,钐钕:sm
x
nd
1-x
,其中0《x《1,钐镨:sm
x
pr
1-x
,其中0《x《1,铕钕:eu
x
nd
1-x
,其中0《x《1,铕铺:eu
x
pr
1-x
,其中0《x《1;b位为单一镍元素或镍与其他过渡金属的组合,优选镍:ni,镍锶:ni
x
sr
1-x
,其中0《x《1,镍锰:ni
x
mn
1-x
,其中0《x《1,镍锌:ni
x
zn
1-x
,其中0《x《1,镍钴:ni
x
co
1-x
,其中0《x《1,镍铁:ni
x
fe
1-x
,其中0《x《1,镍铜:ni
x
cu
1-x
,其中0《x《1,镍钒:ni
xv1-x
,其中0《x《1。更进一步,所述稀土镍基钙钛矿氧化物包括多种不同的形态,稀土镍基钙钛矿氧化物粉末,稀土镍基钙钛矿氧化物纳米线,稀土镍基钙钛矿氧化物薄膜,稀土镍基钙钛矿氧化物陶瓷。在一优选例中,通过稀土元素的改变,改变了氢致相变的速率;在一优选例中,通过稀土镍基钙钛矿氧化物形态的改变改变了氢致相变的速率;在另一优选例中,通过b位元素的掺杂改变了氢致相变的速率。
11.进一步地,所选金属电极需要满足以下条件:1)金属电极的功函数和稀土镍基氧化物材料具有一定的匹配性,当金属电极和稀土镍基氧化物材料的功函数接近时,优选差异在0.1ev以内,本发明提供的触发氢致相变方法的速率缓慢,随着金属电极和稀土镍基氧化物材料的功函数差异增大,本发明提供的触发氢致相变方法的速率增大;
12.2)金属电极在酸性或碱性的化学环境中不会在金属电极表面形成钝化膜。在满足以上两个条件的情况下,当ph值在0-14之间时,优选cu、ni、zn、al、ag、fe、co、pt、pd、mn、ti、cr、ta、w。在一优选例中,pt金属电极的功函数与稀土镍基氧化物接近,优选差异在0.1ev以内,氢致相变速率慢;在一优选例中,cu金属电极的功函数与稀土镍基氧化物相差约0.8ev,
氢致相变速率比pt金属电极大;在另一优选例中,zn金属电极的功函数与稀土镍基氧化物相差超过1ev,氢致相变速率比cu金属电极大。
13.进一步地,所述金属电极的形状可以分为非连续形状和连续形状,其中非连续阵列式金属电极的尺寸和间隔可以从微米到毫米之前改变,非连续阵列式金属电极的形状包括:阵列式的点状、条状、三角形、正方形、五边形和六边形;连续金属电极尺寸为毫米级及以上,连续形状的金属电极则是连续覆盖薄膜表面部分范围,而没有形成阵列式分布结构,包括圆形、矩形、正方形、三角形、五边形、六边形;所述稀土镍基钙钛矿氧化物和金属电极的接触方式主要包括两种形式,第一,金属电极放置在稀土镍基钙钛矿氧化物上,操作简单且通常可分离;第二,将金属电极通过磁控溅射或蒸镀沉积在稀土镍基钙钛矿氧化物上,一般难以将金属电极和稀土镍基钙钛矿氧化物分开。在一优选例中,选用非连续阵列式金属电极;在另一优选例中,选用连续金属电极。
14.进一步地,所述的酸性或碱性化学环境包括溶液环境与气溶胶环境两种情况:酸性或碱性化学溶液环境:1)酸溶液:如盐酸、醋酸、草酸和硫酸,2)酸式盐溶液:如碳酸氢钠、碳酸氢钙、硫酸氢钠、硫氢化钠,3)碱溶液:如氢氧化钠、氢氧化钾、氨水。酸性或碱性气溶胶环境:1)酸性气溶胶环境:氯化氢、硫化氢、二氧化硫、三氧化硫、二氧化碳、二氧化氮、氯气,2)碱性气溶胶环境:氨气、联氨、磷化氢。在没有特殊说明的情况下,以上酸性或碱性气溶胶环境的ph值均位于0-14之间。在一优选例中,是在酸性化学溶液环境中进行亚稳相稀土镍基氧化物氢致相变速率的调控;在一优选例中,酸性化学溶液ph值的改变,将改变亚稳相稀土镍基氧化物氢致相变速率;在一优选例中,是在碱性化学溶液环境中进行亚稳相稀土镍基氧化物氢致相变速率的调控;在一优选例中,是在酸性气溶胶环境中进行亚稳相稀土镍基氧化物氢致相变速率调控;在一优选例中,是在碱性气溶胶环境中进行亚稳相稀土镍基氧化物氢致相变速率调控
15.进一步地,所涉及的亚稳相稀土镍基氧化物氢致电子相变动力学调控过程可以进一步通过电场进行速率和可逆性的调控。当给稀土镍基氧化物施加电位为负的电场或脉冲电场时,可以加速稀土镍基氧化物氢致相变速率;当给稀土镍基钙钛矿氧化材料施加电位为正的电场或脉冲电场时,可以使稀土镍基钙钛矿氧化物材料的氢致相变速率减慢。调整电压的大小,可以调控加快或抑制氢致相变速率的能力,施加的最低电压为毫伏级,最高电压一般不超过一百伏。在一优选例中,通过施加电压的正负调控了该方法触发氢致相变的速率;在另一优选例中,通过改变电压的大小调控该方法触发氢致相变的速率。
16.如上所述亚稳相稀土镍基氧化物氢致电子相变动力学调控的应用,其特征在于,所述应用领域包括两方面:一方面,利用金属电极和稀土镍基氧化物功函数之间的合理匹配,优选差异在0.1ev以内,结合化学环境酸性或碱性程度的选择,可以实现对稀土镍基氧化物氢致电子相变程度和速率的精准设计,氢致相变速率的调控范围为每分钟0.001倍到每分钟1000000倍,从而辅助其在类脑计算、仿生神经等领域内的应用;另一方面,基于金属电极和稀土镍基氧化物在不同ph值的化学环境中氢致电子相变特性的变化规律,可对不同酸碱浓度的化学环境进行探测。在一优选例中,实现了15分钟内,氢致相变速率约为每分钟0.004倍;在另一优选例中,实现了45分钟内氢致相变速率每分钟超过26万倍的增加。
17.本发明人经过广泛而深入的研究,设计了一种亚稳相稀土镍基氧化物氢致电子相变动力学调控方法,其主要构思在于利用稀土镍基钙钛矿氧化物与金属电极间功函数差异
和化学环境ph值的协同作用实现稀土镍基氧化物氢致电子相变程度与速率的动力学精准调控。将一定形状大小的金属电极和稀土镍基氧化物相接触并静置在酸性化学环境中,基于金属和稀土镍基氧化物材料功函数间的差异,驱使溶液中的氢离子发生转移,从而实现稀土镍基氧化物材料的电子构型以一定速率由以三价镍为主导的巡游态电子结构向以二价镍为主导的局域态电子结构转变,并触发材料电阻率的急剧增加;当特定金属电极以可控形状与稀土镍基氧化物材料相接触,并放置在碱性化学环境中,将触发材料电阻率的急剧减小。这一过程还可以结合外加电场的方式加快或可逆稀土镍基氧化物氢致电子相变过程,进一步对稀土镍基钙钛矿氧化物氢致相变速率进行精确调控。这一新的氢致相变触发方法不仅可以实现对稀土镍基氧化物氢致电子相变程度和速率的精准设计,而且也可实现对不同ph值化学环境的检测。
18.与传统利用贵金属pt或pd在高温下氢气退火或电解质溶液中施加电场从而实现氢致相变的方法相比,本发明所提供的氢致相变触发技术与方法,无需氢气气氛,安全性高;无需使用贵金属,成本低;无须加入或施加电场。另一方面,通过金属电极功函数的设计和酸性或碱性化学环境的选择协同触发稀土镍基氧化物的氢致相变,具有精准调控氢致相变程度和速率的特点,可以进一步拓展稀土镍基氧化物在化学溶液酸性或碱性的检测和酸碱气体的监测等领域内的应用。
附图说明
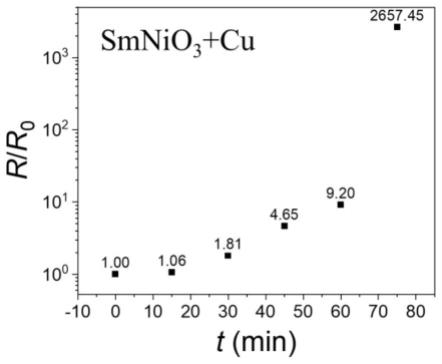
19.图1cu和smnio3薄膜相接触时在0.05mol/l的草酸溶液中的电阻变化随时间变化,其中r0为smnio3薄膜初始电阻,
20.图2cu和ndnio3薄膜相接触时在0.05mol/l的草酸溶液中的电阻变化随时间变化,其中r0为ndnio3薄膜初始电阻,
21.图3pt和smnio3薄膜相接触时在0.05mol/l的草酸溶液中的电阻变化随时间变化,其中r0为smnio3薄膜初始电阻,
22.图4zn和smnio3薄膜相接触时在0.05mol/l的草酸溶液中的电阻变化随时间变化其中,r0为smnio3薄膜初始电阻,
23.图5zn和ndnio3薄膜相接触时在0.05mol/l的草酸溶液中的电阻变化随时间变化,其中r0为ndnio3薄膜初始电阻,
24.图6zn和smnio3薄膜相接触时在0.05mol/l的氢氧化钾溶液中的电阻变化随时间变化,其中r0为smnio3薄膜初始电阻,
25.图7cu和ndnio3薄膜相接触时在0.005mol/l的草酸溶液中的电阻变化随时间变化,其中r0为ndnio3薄膜初始电阻。
具体实施方式
26.如无具体说明,本发明的各种原料均可以通过市售得到;或根据本领域的常规方法制备得到。除非另有定义或说明,本文中所使用的所有专业与科学用语与本领域技术熟练人员所熟悉的意义相同。此外任何与所记载内容相似或均等的方法及材料皆可应用于本发明方法中。
27.本发明的其他方面由于本文的公开内容,对本领域的技术人员而言是显而易见
的。
28.下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件进行。
29.测试方法:我们利用4200半导体表征系统对稀土镍基氧化物的电阻进行测量表征。所述表征方法根据本领域的通用标准进行。
30.实施例1:
31.选用smnio3薄膜为研究对象,测量该薄膜氢致相变发生变化前的电阻或电阻率,称为初始电阻或电阻率,之后将smnio3薄膜与金属cu相接触,并放入0.05mol/l的草酸溶液中,放置一段时间,取出样品并用去离子水冲洗并烘干,然后再次测量该smnio3薄膜的电阻或电阻率,用得到的电阻和初始电阻的比值作为纵坐标,而用时间作为横坐标作图。重复上述过程而得到电阻比值随时间的变化,如图1所示,可以看到电阻比值在15分钟内增大1.06倍,30分钟内增大1.81倍,45分钟内增大4.65倍,60分钟内增大9.20倍,75分钟内增大5657.45倍,氢致相变速率逐渐提高。
32.实施例2:
33.选用ndnio3薄膜为研究对象,测量该薄膜氢致相变发生变化前的电阻或电阻率,称为初始电阻或电阻率,之后将ndnio3薄膜与金属cu相接触,并放入0.05mol/l的草酸溶液中,放置一段时间,取出样品并用去离子水冲洗并烘干,然后再次测量该ndnio3薄膜的电阻或电阻率,用得到的电阻和初始电阻的比值作为纵坐标,而用时间作为横坐标作图。并且可以重复上述过程而得到电阻比值随时间的变化,如图2所示,可以看到电阻比值在15分钟增大10.38倍,在30分钟增大13.63倍,在45分钟增大146.42倍,在60分钟内增大3.64
×
107倍。与实施例1中的图对比表明,在相同时间内,ndnio3的电阻比值随时间增大更多,说明稀土离子的改变,可以调控氢致相变的速率。
34.实施例3:
35.选用smnio3陶瓷为研究对象,测量该陶瓷材料氢致相变发生变化前的电阻或电阻率,称为初始电阻或电阻率,之后将smnio3陶瓷与金属cu相接触,并放入0.05mol/l的草酸溶液中,放置一段时间,取出样品并用去离子水冲洗并烘干,然后再次测量该smnio3陶瓷的电阻或电阻率,用得到的电阻和初始电阻的比值,比值增大说明smnio3陶瓷发生了氢致电子相变,比值随时间增大,说明氢致相变速率增大。与实施例1相比,比值随时间增大的速率越快,说明氢致相变的速率越快。
36.实施例4:
37.选用smni
0.5
cu
0.5
o3薄膜为研究对象,测量该薄膜氢致相变发生变化前的电阻或电阻率,称为初始电阻或电阻率,之后将smni
0.5
cu
0.5
o3薄膜与金属cu相接触,并放入0.05mol/l的草酸溶液中,放置一段时间,取出样品并用去离子水冲洗并烘干,然后再次测量该smni
0.5
cu
0.5
o3薄膜的电阻或电阻率,与初始电阻或电阻率的比值随时间发生明显增大,说明稀土镍基钙钛矿氧化物与金属电极间功函数差异和化学环境ph值的协同作用可以触发smni
0.5
cu
0.5
o3薄膜的氢致相变,并且比值增加越快,说明氢致相变速率越快。与实施例1相比,相同时间内,比值增大越快的稀土镍基氧化物,说明氢致相变速率越快。
38.实施例5:
39.选用smnio3薄膜为研究对象,测量该薄膜氢致相变发生变化前的电阻或电阻率,称为初始电阻或电阻率,之后将smnio3薄膜与金属pt相接触,并放入0.05mol/l的草酸溶液中,放置一段时间,取出样品并用去离子水冲洗并烘干,然后再次测量该smnio3薄膜的电阻或电阻率,用得到的电阻和初始电阻的比值作为纵坐标,而用时间作为横坐标作图。重复上述过程而得到电阻比值随时间的变化,如图3所示,可以看到电阻比值在15分钟内增大0.99倍,30分钟内增大1.05倍,45分钟内增大1.13倍,60分钟内增大1.31倍,电阻比值随时间变化不明显,氢致相变几乎不发生。与实施例1相比,相同时间内,pt作为金属电极时触发氢致相变的速率非常低。
40.实施例6:
41.选用smnio3薄膜为研究对象,测量该薄膜氢致相变发生变化前的电阻或电阻率,称为初始电阻或电阻率,之后将smnio3薄膜与金属zn相接触,并放入0.05mol/l的草酸溶液中,放置一段时间,取出样品并用去离子水冲洗并烘干,然后再次测量该smnio3薄膜的电阻或电阻率,用得到的电阻和初始电阻的比值作为纵坐标,而用时间作为横坐标作图。并且可以重复上述过程而得到电阻比值随时间的变化,如图4所示,电阻比值15分钟内增大1.28倍,30分钟内增大23.19倍,45分钟内增大155.31倍,60分钟内增大1659844.27倍,氢致相变速率随时间增加,并且与实施例1中的图对比可以发现,选择金属zn作为电极时,smnio3薄膜的电阻比值在相同的时间内变化更多,说明金属zn电极触发的smnio3薄膜的氢致相变速率更快,说明选择不同的金属电极可以调控稀土镍基钙钛矿氧化物材料具有不同的氢致相变速率。
42.实施例7:
43.选用smnio3薄膜为研究对象,测量该薄膜氢致相变发生变化前的电阻或电阻率,称为初始电阻或电阻率,之后在两个smnio3薄膜上分别沉积阵列式圆形和三角形cu金属电极,其中圆形cu金属电极的直径和间隔都为100微米,而三角形cu金属电极的变长为100微米,间隔也为100微米,并分别放入0.05mol/l的草酸溶液中,放置一段时间,取出样品并用去离子水冲洗并烘干,然后再次测量两个smnio3薄膜的电阻或电阻率,用得到的电阻和初始电阻的比值作为纵坐标,而用时间作为横坐标作图。并且可以重复上述过程而得到电阻比值随时间的变化。两者对比发现,圆形和三角形cu金属电极阵列都可以使得smnio3薄膜的电阻比值随时间增大而增大,说明不同形状的金属电极都可以触发smnio3薄膜的氢致电子相变,且氢致相变的速率随时间增大而增大。
44.实施例8:
45.选用smnio3薄膜为研究对象,测量该薄膜氢致相变发生变化前的电阻或电阻率,称为初始电阻或电阻率,之后在smnio3薄膜沉积1毫米乘以1毫米的矩形连续cu金属电极,并分别放入0.05mol/l的草酸溶液中,放置一段时间,取出样品并用去离子水冲洗并烘干,然后再次测量smnio3薄膜的电阻或电阻率,用得到的电阻和初始电阻的比值作为纵坐标,而用时间作为横坐标作图。并且可以重复上述过程而得到电阻比值随时间的变化。smnio3薄膜的电阻比值随时间增大而增大,说明连续矩形cu金属电极可以触发这种情况下smnio3薄膜的氢致电子相变,且氢致相变速率随时间增大而增大。但和实施例7中的阵列式cu金属电极相比,氢致相变速率要更小。
46.实施例9:
47.选用ndnio3薄膜为研究对象,测量该薄膜氢致相变发生变化前的电阻或电阻率,称为初始电阻或电阻率,之后将ndnio3薄膜与金属zn相接触,并放入0.05mol/l的草酸溶液中,放置一段时间,取出样品并用去离子水冲洗并烘干,然后再次测量该ndnio3薄膜的电阻或电阻率,用得到的电阻和初始电阻的比值作为纵坐标,而用时间作为横坐标作图。并且可以重复上述过程而得到电阻比值随时间的变化,如图4所示,可以看到电阻比值随时间而增大,在15分钟内,电阻比值增大146.57倍,在30分钟内,电阻比值增大2064772.13倍,在45分钟内,电阻比值增大1.19
×
107倍,说明氢致相变速率随时间增加。且与实施例2中的结果对比可以明显发现,金属zn电极在相同的时间内能够使得ndnio3薄膜的电阻比值增加更快,说明此时氢致电子相变速率更快。
48.实施例10:
49.选用smnio3薄膜为研究对象,测量该薄膜氢致相变发生变化前的电阻或电阻率,称为初始电阻或电阻率,之后将两个smnio3薄膜与金属al相接触,并分别放入到15%的二氧化硫和二氧化碳的气溶胶环境中,放置一段时间,取出样品并用去离子水冲洗并烘干,然后再次测量该smnio3薄膜的电阻或电阻率,用得到的电阻和初始电阻的比值作为纵坐标,而用时间作为横坐标作图。并且可以重复上述过程而得到电阻比值随时间的变化,两种情况下smnio3薄膜的电阻比值随时间而增大,但是在二氧化硫气溶胶环境中的smnio3薄膜的电阻率增加更快,说明此时smnio3薄膜的氢致电子相变速率更快,进一步证明smnio3薄膜可以用于酸性气溶胶化学环境的检测。
50.实施例11:
51.选用smnio3薄膜为研究对象,测量该薄膜氢致相变发生变化前的电阻或电阻率,称为初始电阻或电阻率,之后将smnio3薄膜与金属zn相接触,并放入0.05mol/l的氢氧化钾溶液中,放置一段时间,取出样品并用去离子水冲洗并烘干,然后再次测量该smnio3薄膜的电阻或电阻率,用得到的电阻和初始电阻的比值作为纵坐标,而用时间作为横坐标作图。并且可以重复上述过程而得到电阻比值随时间的变化,如图6所示,可以看到电阻比值随时间而减小,在15分钟内,电阻减小为初始电阻的0.98倍,在30分钟内,电阻减小为初始电阻的0.96倍,在45分钟内,电阻减小为初始电阻的0.92倍,在60分钟内电阻减小为初始电阻的0.57倍,在75分钟内电阻减小为初始电阻的0.50倍,之后90分钟到120分钟内电阻保持不变,以上说明在碱性条件下smnio3薄膜的氢致电子相变被抑制。
52.实施例12:
53.选用smnio3薄膜为研究对象,测量该薄膜氢致相变发生变化前的电阻或电阻率,称为初始电阻或电阻率,之后将两个smnio3薄膜与金属al相接触,并分别放入到15%的氨气气溶胶环境中,放置一段时间,取出样品并用去离子水冲洗并烘干,然后再次测量该smnio3薄膜的电阻或电阻率,用得到的电阻和初始电阻的比值作为纵坐标,而用时间作为横坐标作图。并且可以重复上述过程而得到电阻比值随时间的变化,smnio3薄膜的电阻比值随时间而减小,说明此时smnio3薄膜的氢致电子相变被抑制,进一步证明smnio3薄膜可以用于碱性气溶胶化学环境的检测。
54.实施例13:
55.选用ndnio3薄膜为研究对象,测量该薄膜氢致相变发生变化前的电阻或电阻率,称为初始电阻或电阻率,之后将ndnio3薄膜与金属cu相接触,并放入0.005mol/l的草酸溶
液中,放置一段时间,取出样品并用去离子水冲洗并烘干,然后再次测量该ndnio3薄膜的电阻或电阻率,用得到的电阻和初始电阻的比值作为纵坐标,而用时间作为横坐标作图。并且可以重复上述过程而得到电阻比值随时间的变化,如图7所示,可以看到,在15分钟内,电阻比值增大1.23倍,在30分钟内,电阻比值增大9.87倍,45分钟内,电阻比值增大6478.63倍,60分钟内,电阻比值增大8328倍,75分钟内,电阻比值增大112347.62倍,在90分钟内,电阻比值增大87305.77倍,105分钟内,电阻比值增大543275.51倍。与实施例2中的结果对比,可以发现,在相同的条件下,ph值不同时,氢致电子相变速率不同,说明ph值可以调控ndnio3薄膜的氢致电子相变速率。
56.实施例14:
57.选用ndnio3薄膜为研究对象,测量该薄膜氢致相变发生变化前的电阻或电阻率,称为初始电阻或电阻率,之后将ndnio3薄膜与金属cu相接触,并放入0.0005mol/l的草酸溶液中,放置一段时间,取出样品并用去离子水冲洗并烘干,然后再次测量该ndnio3薄膜的电阻或电阻率,用得到的电阻和初始电阻的比值作为纵坐标,而用时间作为横坐标作图。并且可以重复上述过程而得到电阻比值随时间的变化,电阻比值随时间增大而增大,与实施例2中的结果对比,可以发现,在相同的条件下,ph值不同时,氢致电子相变速率不同,说明ph值可以调控ndnio3薄膜的氢致电子相变速率。
58.实施例15:
59.选用sm
0.5
nd
0.5
nio3薄膜为研究对象,测量该薄膜氢致相变发生变化前的电阻或电阻率,称为初始电阻或电阻率,之后将sm
0.5
nd
0.5
nio3薄膜与金属cu相接触,并放入0.05mol/l的草酸溶液中,放置一段时间,取出样品并用去离子水冲洗并烘干,然后再次测量该sm
0.5
nd
0.5
nio3薄膜的电阻或电阻率,与初始电阻或电阻率的比值随时间发生明显增大,说明稀土镍基钙钛矿氧化物与金属电极间功函数差异和化学环境ph值的协同作用可以触发sm
0.5
nd
0.5
nio3薄膜的氢致相变,并且与实施例1相比,电阻比值增加越快,说明氢致相变速率越快。
60.实施例16:
61.选用smnio3薄膜为研究对象,测量该薄膜氢致相变发生变化前的电阻或电阻率,称为初始电阻或电阻率,之后将smnio3薄膜与金属cu相接触,并放入0.05mol/l的硫酸溶液中,放置一段时间,取出样品并用去离子水冲洗并烘干,然后再次测量该smnio3薄膜的电阻或电阻率,用得到的电阻和初始电阻的比值作为纵坐标,而用时间作为横坐标作图。并且可以重复上述过程而得到电阻比值随时间的变化,电阻比值随时间而增大,说明smnio3薄膜发生了氢致电子相变,且与实施例1对比,可以发现,相同时间内,此时的氢致电子相变速率更快。
62.实施例17:
63.选用smnio3薄膜为研究对象,测量该薄膜氢致相变发生变化前的电阻或电阻率,称为初始电阻或电阻率,之后将smnio3薄膜与金属cu相接触,并放入0.05mol/l草酸溶液中,并给smnio3薄膜施加-0.1v的负电位,放置一段时间,取出样品并用去离子水冲洗并烘干,然后再次测量该smnio3薄膜的电阻或电阻率,用得到的电阻和初始电阻的比值作为纵坐标,而用时间作为横坐标作图。与实施例1的结果对比,可以发现在给smnio3薄膜施加负电场时,电阻比值增大速率明显提高,说明施加电场可以进一步加速smnio3薄膜氢致电子
相变速率。
64.实施例18:
65.选用smnio3薄膜为研究对象,测量该薄膜氢致相变发生变化前的电阻或电阻率,称为初始电阻或电阻率,之后将smnio3薄膜与金属cu相接触,并放入0.05mol/l草酸溶液中,并给smnio3薄膜施加0.1v的正电位,放置一段时间,取出样品并用去离子水冲洗并烘干,然后再次测量该smnio3薄膜的电阻或电阻率,用得到的电阻和初始电阻的比值作为纵坐标,而用时间作为横坐标作图。与实施例1的结果对比,可以发现在给smnio3薄膜施加正向电场时,电阻比值增大速率明显减慢,说明施加电场可以进一步抑制smnio3薄膜氢致电子相变速率。
66.实施例19:
67.选用smnio3薄膜为研究对象,测量该薄膜氢致相变发生变化前的电阻或电阻率,称为初始电阻或电阻率,之后将smnio3薄膜与金属cu相接触,并放入0.05mol/l草酸溶液中,并给smnio3薄膜施加-1v的负电位,放置一段时间,取出样品并用去离子水冲洗并烘干,然后再次测量该smnio3薄膜的电阻或电阻率,用得到的电阻和初始电阻的比值作为纵坐标,而用时间作为横坐标作图。与实施例17的结果对比,可以发现在给smnio3薄膜施加负向电场时,电阻比值增大速率进一步提升,说明施加更大的负电场可以进一步加速smnio3薄膜氢致电子相变速率。
68.实施例20:
69.选用smnio3薄膜为研究对象,测量该薄膜氢致相变发生变化前的电阻或电阻率,称为初始电阻或电阻率,之后将smnio3薄膜与金属cu相接触,并放入0.05mol/l草酸溶液中,并给smnio3薄膜施加1v的正向电位,放置一段时间,取出样品并用去离子水冲洗并烘干,然后再次测量该smnio3薄膜的电阻或电阻率,用得到的电阻和初始电阻的比值作为纵坐标,而用时间作为横坐标作图。与实施例18的结果对比,可以发现在给smnio3薄膜施加正向电场时,电阻比值增大速率进一步减慢,说明施加更大的正向电场可以进一步抑制smnio3薄膜氢致电子相变速率。
70.实施例21:
71.选用smnio3薄膜为研究对象,测量该薄膜氢致相变发生变化前的电阻或电阻率,称为初始电阻或电阻率,之后将smnio3薄膜与金属zn相接触,并放入0.05mol/l的氨水溶液中,放置一段时间,取出样品并用去离子水冲洗并烘干,然后再次测量该smnio3薄膜的电阻或电阻率,用得到的电阻和初始电阻的比值作为纵坐标,而用时间作为横坐标作图。并且可以重复上述过程而得到电阻比值随时间的变化,电阻比值随时间而减小,与实施例11相比,电阻比值随时间减少的速率减慢,说明在弱碱性条件下smnio3薄膜的氢致电子相变抑制能力减弱。
72.以上所述仅为本发明的较佳实施例而已,并非用以限定本发明的实质技术内容范围,本发明的实质技术内容是广义地定义于申请的权利要求范围中,任何他人完成的技术实体或方法,若是与申请的权利要求范围所定义的完全相同,也或是一种等效的变更,均将被视为涵盖于该权利要求范围之中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1