中药组合物的质量检测方法与流程
1.本发明涉及医药技术领域,特别是涉及一种中药组合物的质量检测方法。
背景技术:
2.有一种主要由大黄、黄芪、桑白皮、苦参、党参、白术、茯苓、制何首乌、白芍、丹参、川芎、菊花、姜半夏、车前草、柴胡和甘草制备而成的中药组合物,其具有通腑降浊、健脾利湿、活血化瘀的功效,可用于慢性肾功能衰竭,氮质血症期和尿毒症早期、中医辨证属脾虚湿浊症和脾虚血瘀症者。
3.该中药组合物中,柴胡、茯苓、丹参等具有突出的抗炎、清除自由基作用,利尿降血糖作用显著,可缓解或预防肾病的进一步恶化,降低尿蛋白、微量白蛋白,清除血液中的ages,红细胞数明显增加,改善肾性贫血,清除维持性血液透析(mhd)患者血浆中crp、白介素和肿瘤坏死因子等炎症因子。郭陆晋通过对病人为期4周的对症连续服药,发现治疗组在尿蛋白,血脂,肾功能等指标比对照组有显著改善,说明尿毒清颗粒能改善患者高血压所致的肾损害。苗绪红等通过灌胃腺嘌呤诱导大鼠形成慢性肾功能衰竭,检测大鼠肾皮质转化生长因子-β1(transfer growth factor-β1,tgf-β1)及其下游基因mrna的表达变化,结果发现,该制剂治疗后,大鼠肾脏形态有所改善;血肌酐,尿素氮水平下降;大鼠tgf-β1及其下游基因mrna表达减少,tgf-β1蛋白表达减少。对于丹酚酸b的药理作用研究,国内外有大量的文献记载,包括孙仁弟等对丹酚酸b的心脑血管保护,肝肾保护,抗纤维化等多种药理活性进行了详尽的研究。丹酚酸b除了神经保护以及抗氧化等作用外,对于高糖(hg)诱导的氧化应激诱导下,线粒体途径的激活和雪旺氏细胞(sc)的体外细胞凋亡均有保护作用。
4.可见,该中药组合物的临床疗效可靠。然而,目前对该中药组合物的质量研究还不够完善,有必要对其进行全面质量控制。
技术实现要素:
5.基于此,本发明提供一种中药组合物的质量检测方法,可使中药组合物在生产和使用中得到有效的产品质量保证,进而保障药品疗效。
6.具体技术方案如下:
7.一种中药组合物的质量检测方法,包括以下步骤:
8.取待测中药组合物样品,其中,中药组合物的理论制备原料包括大黄、黄芪、桑白皮、苦参、党参、白术、茯苓、制何首乌、白芍、丹参、川芎、菊花、姜半夏、车前草、柴胡和甘草;
9.采用薄层色谱法分别检测所述待测中药组合物样品中是否含有大黄、制何首乌、黄芪、丹参、苦参、柴胡、桑白皮、茯苓、党参、甘草和白芍;
10.采用高效液相色谱法分别检测所述待测中药组合物样品中芍药苷、丹酚酸b、黄芪甲苷、2,3,5,4
’‑
四羟基二苯乙烯-2-o-β-d葡萄糖苷和苦参碱的含量,结合含量确定待测中药组合物样品的质量;
11.对所述待测中药组合物样品进行性状检测。
12.可选地,所述含量同时满足以下条件时,确定待测中药组合物样品的质量为合格:
13.a)所述芍药苷的含量不少于0.7mg/g;
14.b)所述丹酚酸b的含量不少于0.25mg/g;
15.c)所述黄芪甲苷的含量不少于0.1mg/g;
16.d)所述2,3,5,4
’‑
四羟基二苯乙烯-2-o-β-d葡萄糖苷的含量不少于0.1mg/g;
17.e)所述苦参碱的含量不少于0.3mg/g。
18.可选地,采用薄层色谱法检测所述待测中药组合物样品中是否含有大黄和制何首乌的方法包括以下步骤:
19.取所述待测中药组合物样品,加硫酸溶液加热回流,放冷后,加氯仿加热回流,取氯仿液,干燥,残渣加氯仿溶解,制备供试品溶液;
20.制备大黄对照药材溶液和制何首乌对照药材溶液;制备含有大黄酸、大黄素和大黄素甲醚的混合对照品溶液;
21.分别吸取所述供试品溶液、大黄对照药材溶液、制何首乌对照药材溶液以及混合对照品溶液,点于同一硅胶g或h薄层板上,以体积比为(10~20):(3~7):(0.5~2)的石油醚、甲酸乙酯和甲酸的上层溶液为展开剂,展开,晾干后观察。
22.优选地,每3g~7g所述待测中药组合物样品对应加入2mol/l~5mol/l的硫酸溶液20ml~50ml。更优选地,每5g所述待测中药组合物样品对应加入3mol/l的硫酸溶液20ml~40ml。
23.优选地,加硫酸溶液加热回流的时间为0.5小时~2小时。更优选地,加硫酸溶液加热回流的时间为0.5小时~1.5小时。
24.优选地,每3g~7g所述待测中药组合物样品对应加入氯仿20ml~50ml。更优选地,每5g所述待测中药组合物样品对应加入氯仿20ml~40ml。
25.优选地,加氯仿加热回流的时间为0.5小时~2小时。更优选地,加氯仿加热回流的时间为0.5小时~1.5小时。
26.优选地,制备大黄对照药材溶液的方法包括:取大黄对照药材,参照上述制备供试品溶液的方法,制备大黄对照药材溶液。更优选地,每0.5g~2g所述大黄对照药材对应加入2mol/l~5mol/l的硫酸溶液20ml~50ml。对应加入氯仿20ml~50ml。
27.优选地,制备制何首乌对照药材溶液的方法包括:取制何首乌对照药材,参照上述制备供试品溶液的方法,制备制何首乌对照药材溶液。更优选地,每0.5g~2g所述何首乌对照药材对应加入2mol/l~5mol/l的硫酸溶液20ml~50ml。对应加入氯仿20ml~50ml。
28.优选地,制备含有大黄酸对照品、大黄素对照品和大黄素甲醚对照品的混合对照品溶液的溶剂为甲醇。更优选地,混合对照品溶液中,大黄酸对照品的浓度为0.5mg/ml~2mg/ml,大黄素对照品的浓度为0.5mg/ml~2mg/ml,大黄素甲醚对照品的浓度为0.5mg/ml~2mg/ml。
29.优选地,以体积比为(14~16):(4~6):(0.5~1.5)的石油醚、甲酸乙酯和甲酸的上层溶液为展开剂。
30.可选地,采用薄层色谱法检测所述待测中药组合物样品中是否含有黄芪的方法包括以下步骤:
31.取所述待测中药组合物样品,加正丁醇加热回流,滤过,取滤液,进行碱洗,弃去碱
液,加水饱和的正丁醇洗至中性,取正丁醇提取液干燥,残渣加甲醇溶解,制备供试品溶液;
32.制备黄芪甲苷对照品溶液;
33.分别吸取所述供试品溶液和黄芪甲苷对照品溶液,点于同一硅胶g或h薄层板上,以体积比为(12~14):(6~8):(1~4)的氯仿、甲醇和水的下层溶液为展开剂,展开,晾干后观察。
34.优选地,每3g~7g所述待测中药组合物样品对应加入正丁醇10ml~50ml。更优选地,每5g所述待测中药组合物样品对应加入正丁醇10ml~30ml。
35.优选地,加正丁醇加热回流的时间为1小时~3小时。
36.优选地,所述碱液为氢氧化钠溶液。更优选地,所述氢氧化钠溶液的质量分数为0.5%~2%。更优选地,所述氢氧化钠溶液的质量分数为0.5%~1.5%。
37.优选地,碱洗的次数为2~5次。更优选地,碱洗的次数为2~4次。
38.更优选地,每次碱洗时,每3g~7g所述待测中药组合物样品对应加入质量分数为0.5%~2%氢氧化钠溶液10ml~50ml。更优选地,每次碱洗时,每5g所述待测中药组合物样品对应加入质量分数为0.5%~1.5%氢氧化钠溶液10ml~20ml。
39.优选地,制备黄芪甲苷对照品溶液的溶剂为甲醇。更优选地,对照品溶液中,黄芪甲苷对照品的浓度为0.5mg/ml~2mg/ml。
40.优选地,以体积比为(12~14):(6~8):(1~3)的氯仿、甲醇和水的下层溶液为展开剂。
41.优选地,晾干后,还包括喷以体积分数为5%~20%的硫酸的乙醇溶液,加热至斑点显色清晰的步骤。优选地,晾干后,还包括喷以质量分数为5%~15%的硫酸的乙醇溶液,加热至斑点显色清晰的步骤。
42.可选地,采用薄层色谱法检测所述待测中药组合物样品中是否含有丹参的方法包括以下步骤:
43.取所述待测中药组合物样品,加水溶解后滤过,取滤液,加乙醚提取,取乙醚相干燥,残渣加乙醇溶解,制备供试品溶液;
44.制备丹参对照药材溶液和原儿茶醛对照品溶液;
45.分别吸取所述供试品溶液、丹参对照药材溶液和原儿茶醛对照品溶液,点于同一硅胶g或h薄层板上,以体积比为(1~3):(3~5):(3~5):(0.3~0.7)的己烷、苯、乙酸乙酯和甲酸作为展开剂,展开,晾干后观察。
46.优选地,每3g~7g所述待测中药组合物样品对应加入水20ml~100ml。更优选地,每5g所述待测中药组合物样品对应加入水40ml~60ml。
47.优选地,加乙醚提取次数为1~3次。更优选地,每次提取时,每3g~7g所述待测中药组合物样品对应加入乙醚10ml~50ml。更优选地,每次提取时,每5g所述待测中药组合物样品对应加入乙醚10ml~20ml。
48.优选地,制备丹参对照药材溶液的方法包括:取丹参对照药材,加水煎煮,将所得煎液浓缩,取浓缩液,参照上述制备供试品溶液的方法,制备丹参对照药材溶液。
49.更优选地,加水煎煮的次数为1~3次。更优选地,加水煎煮的时间为5分钟~60分钟。更优选地,加水煎煮的时间为5分钟~15分钟。更优选地,每3g~7g丹参对照药材对应浓缩至5ml~15ml。更优选地,每4ml~6ml浓缩液,对应加入水20ml~100ml,对应加入乙醚
10ml~50ml。
50.优选地,制备原儿茶醛对照品溶液的溶剂为乙醇。更优选地,对照品溶液中,原儿茶醛对照品的浓度为0.2mg/ml~5mg/ml。
51.优选地,晾干后,还包括喷以质量分数为0.05%~0.5%的2、4-二硝基苯肼的乙醇溶液的步骤。更优选地,晾干后,还包括喷以质量分数为0.05%~0.15%的2、4-二硝基苯肼的乙醇溶液的步骤。
52.可选地,采用薄层色谱法检测所述待测中药组合物样品中是否含有苦参的方法包括以下步骤:
53.取所述待测中药组合物样品,加氯仿和浓氨溶液,静置后滤过,取滤液干燥,残渣加氯仿溶解,制备供试品溶液;
54.制备苦参碱对照品溶液;
55.分别吸取所述供试品溶液、苦参碱对照品溶液,点于同一硅胶g或h薄层板上,以体积比为(10~30):(10~30):(2~4):(0.5~2)的甲苯、乙酸乙酯、甲醇和浓氨溶液为展开剂,展开,晾干后观察。
56.优选地,每3g~7g所述待测中药组合物样品对应加入氯仿30ml~80ml,加入浓氨溶液0.1ml~0.5ml。更优选地,每5g所述待测中药组合物样品对应加入氯仿40ml~60ml,加入浓氨溶液0.2ml~0.4ml。
57.优选地,每3g~7g所述待测中药组合物样品对应的残渣用0.3ml~1ml氯仿溶解。更优选地,每5g所述待测中药组合物样品对应的残渣用0.4ml~0.6ml氯仿溶解。
58.优选地,制备苦参碱对照品溶液的溶剂为乙醇。更优选地,对照品溶液中,苦参碱对照品的浓度为0.05mg/ml~0.5mg/ml。
59.优选地,以体积比为(15~25):(15~25)):(2~4):(0.5~1.5)的甲苯、乙酸乙酯、甲醇和浓氨溶液为展开剂。
60.优选地,晾干后,还包括喷以稀碘化铋试液的步骤。
61.可选地,采用薄层色谱法检测所述待测中药组合物样品中是否含有柴胡的方法包括以下步骤:
62.取所述待测中药组合物样品,加水超声提取,滤过,取滤液通过ab-8型大孔吸附树脂柱,用体积分数为40%~60%的乙醇溶液洗脱,弃去洗脱液,用体积分数为80%~100%的乙醇溶液继续洗脱,取洗脱液干燥,残渣加甲醇溶解,制备供试品溶液;
63.制备柴胡对照药材溶液;
64.分别吸取所述供试品溶液和柴胡对照药材溶液,点于同一硅胶g或h薄层板上,以体积比为(9~13):(1~3):(0.5~2)的乙酸乙酯、乙醇和水为展开剂,展开,晾干后观察。
65.优选地,每3g~7g所述待测中药组合物样品对应加入水20ml~50ml。优选地,每5g所述待测中药组合物样品对应加入水20ml~40ml。
66.优选地,加水超声提取的时间为10分钟~30分钟。
67.优选地,每3g~7g所述待测中药组合物样品对应加入体积分数为40%~60%的乙醇溶液30ml~70ml,加入体积分数为80%~100%的乙醇溶液30ml~70ml。更优选地,每5g所述待测中药组合物样品对应加入体积分数为45%~55%的乙醇溶液40ml~60ml,加入体积分数为85%~95%的乙醇溶液4ml~6ml。
68.优选地,制备柴胡对照药材溶液的方法包括:取柴胡对照药材,加甲醇适量,超声提取,滤过,滤液浓缩至适量。
69.更优选地,每0.1g~0.5g所述柴胡对照药材对应加入甲醇15ml~25ml。更优选地,超声提取的时间为5min~15min。更优选地,每0.1g~0.5g所述柴胡对照药材对应浓缩至4ml~6ml。
70.优选地,以体积比为(10~13):(1~3):(0.5~1.5)的乙酸乙酯、乙醇和水为展开剂。
71.优选地,晾干后,喷以质量分数为1%~3%的对二甲基甲醛的硫酸水溶液,加热至斑点显色清晰的步骤。更优选地,硫酸水溶液的质量分数为30%~50%。
72.可选地,采用薄层色谱法检测所述待测中药组合物样品中是否含有桑白皮的方法包括以下步骤:
73.取所述待测中药组合物样品,加饱和碳酸钠溶液超声提取,滤过,取滤液,加盐酸调节ph值至1~2,静置后滤过,取滤液,加乙酸乙酯振摇提取,取乙酸乙酯相,干燥,残渣加甲醇溶解,制备供试品溶液;
74.制备桑白皮对照药材溶液;
75.分别吸取所述供试品溶液和柴胡对照药材溶液,点于同一聚酰胺薄层板,以醋酸为展开剂,展开,晾干后观察。
76.优选地,每0.5g~5g所述待测中药组合物样品对应加入饱和碳酸钠溶液10ml~50ml。更优选地,每2g所述待测中药组合物样品对应加入饱和碳酸钠溶液10ml~30ml。
77.优选地,加饱和碳酸钠溶液超声提取的时间为10分钟~30分钟。更优选地,加饱和碳酸钠溶液超声提取的时间为15分钟~25分钟。
78.优选地,静置的时间为20分钟~50分钟。更优选地,静置的时间为25分钟~35分钟。
79.优选地,加乙酸乙酯振摇提取的次数为1~5次。优选地,加乙酸乙酯振摇提取的次数为1~3次。
80.更优选地,每次提取时,每0.5g~5g所述待测中药组合物样品对应加入乙酸乙酯10ml~50ml。更优选地,每次提取时,每2g所述待测中药组合物样品对应加入乙酸乙酯15ml~25ml。
81.优选地,制备桑白皮对照药材溶液的方法包括:取桑白皮对照药材,参照上述制备供试品溶液的方法,制备桑白皮对照药材溶液。更优选地,每0.5g~5g桑白皮对照药材对应加入饱和碳酸钠溶液10ml~50ml。对应加入乙酸乙酯10ml~50ml。
82.可选地,采用薄层色谱法检测所述待测中药组合物样品中是否含有茯苓的方法包括以下步骤:
83.取所述待测中药组合物样品,加水超声提取,滤过,取滤液,加水饱和的正丁醇萃取,取正丁醇相,干燥,残渣加甲醇溶解,制备供试品溶液;
84.制备茯苓对照药材溶液;
85.分别吸取所述供试品溶液和茯苓对照药材溶液,点于同一硅胶g或h薄层板上,以体积比(15~25):(3~7):(0.3~0.7)的甲苯、乙酸乙酯和甲酸为展开剂,展开,晾干后观察。
86.优选地,每3g~7g所述待测中药组合物样品对应加入水20ml~60ml。更优选地,每5g所述待测中药组合物样品对应加入水30ml~50ml。
87.优选地,加水超声提取的时间为20分钟~50分钟。更优选地,加水超声提取的时间为25分钟~35分钟。
88.优选地,加水饱和的正丁醇萃取的次数为2~5次。更优选地,加水饱和的正丁醇萃取的次数为2~4次。
89.更优选地,每次萃取时,每3g~7g所述待测中药组合物样品对应加入水饱和的正丁醇10ml~30ml。更优选地,每次萃取时,每5g所述待测中药组合物样品对应加入水饱和的正丁醇15ml~25ml。
90.优选地,制备茯苓对照药材溶液的方法包括:取茯苓对照药材,参照上述制备供试品溶液的方法,制备茯苓对照药材溶液。更优选地,每0.5g~2g茯苓对照药材对应加入水20ml~60ml。对应加入水饱和的正丁醇10ml~30ml。
91.优选地,以体积比(15~25):(4~6):(0.4~0.6)的甲苯、乙酸乙酯和甲酸为展开剂。
92.优选地,晾干后,还包括喷以质量分数为1~3%香草醛的硫酸溶液-乙醇(3~5:0.5~1.5,v/v)混合溶液,加热至斑点显色清晰的步骤。
93.可选地,采用薄层色谱法检测所述待测中药组合物样品中是否含有党参的方法包括以下步骤:
94.取所述待测中药组合物样品,加水超声提取,滤过,取滤液通过ab-8型大孔吸附树脂柱,用体积分数为40%~60%的乙醇溶液洗脱,弃去洗脱液,用体积分数为80%~100%的乙醇溶液继续洗脱,取洗脱液干燥,残渣加甲醇溶解,制备供试品溶液;
95.制备党参对照药材溶液和党参炔苷对照品溶液;
96.分别吸取所述供试品溶液、党参对照药材溶液和党参炔苷对照品溶液,点于同一硅胶g或h薄层板上,以体积比为(6~8):(0.5~2):(0.3~0.7)的正丁醇、冰醋酸和水为展开剂,展开,晾干后观察。
97.优选地,每3g~7g所述待测中药组合物样品对应加入水20ml~50ml。更优选地,每5g所述待测中药组合物样品对应加入水20ml~40ml。
98.优选地,加水超声提取的时间为10分钟~30分钟。更优选地,加水超声提取的时间为15分钟~25分钟。
99.优选地,每3g~7g所述待测中药组合物样品对应加入体积分数为40%~60%的乙醇溶液30ml~70ml,加入体积分数为80%~100%的乙醇溶液30ml~70ml。更优选地,每5g所述待测中药组合物样品对应加入体积分数为45%~55%的乙醇溶液40ml~60ml,加入体积分数为85%~95%的乙醇溶液40ml~60ml。
100.优选地,制备党参对照药材溶液的方法包括:取党参对照药材,参照上述制备供试品溶液的方法,制备党参对照药材溶液。更优选地,每0.5g~2g党参对照药材对应加入水20ml~50ml。对应加入体积分数为40%~60%的乙醇溶液30ml~70ml,加入体积分数为80%~100%的乙醇溶液30ml~70ml。
101.优选地,制备党参炔苷对照品溶液的溶剂为甲醇。更优选地,对照品溶液中,党参炔苷对照品的浓度为0.5mg/ml~2mg/ml。
102.优选地,以体积比为(6~8):(0.5~1.5):(0.4~0.6)的正丁醇、冰醋酸和水为展开剂。
103.优选地,晾干后,还包括喷以质量分数为5%~15%的硫酸的乙醇溶液,加热至斑点显色清晰的步骤。
104.可选地,采用薄层色谱法检测所述待测中药组合物样品中是否含有甘草和白芍的方法包括以下步骤:
105.取所述待测中药组合物样品,加水溶解,加水饱和的正丁醇萃取,取正丁醇相,干燥,残渣加水溶解,通过c
18
小柱,用水和甲醇洗脱,取甲醇洗脱液,干燥,残渣加甲醇溶解,制备供试品溶液;
106.制备甘草对照药材溶液和芍药苷对照品溶液;
107.分别吸取所述供试品溶液、甘草对照药材溶液和芍药苷对照品溶液,点于同一硅胶g或h薄层板上,以体积比为(13~17):(0.5~2):(0.5~2):(1~3)的乙酸乙酯、甲酸、冰醋酸和水为展开剂,展开,晾干后观察。
108.优选地,每10g~20g所述待测中药组合物样品对应加入水50ml~150ml。更优选地,每15g所述待测中药组合物样品对应加入水90ml~110ml。
109.优选地,加水饱和的正丁醇萃取的次数为1~3次,更优选地,每次萃取时,每10g~20g所述待测中药组合物样品对应加入水饱和的正丁醇30ml~50ml。更优选地,每次萃取时,每15g所述待测中药组合物样品对应加入水饱和的正丁醇35ml~45ml。
110.优选地,用水洗脱时,每10g~20g所述待测中药组合物样品对应加入水10ml~30ml。更优选地,用水洗脱时,每15g所述待测中药组合物样品对应加入水15ml~25ml。
111.优选地,用甲醇洗脱时,每10g~20g所述待测中药组合物样品对应加入甲醇5ml~15ml。更优选地,用甲醇洗脱时,每15g所述待测中药组合物样品对应加入甲醇8ml~12ml。
112.优选地,每10g~20g所述待测中药组合物样品对应的残渣用0.5ml~1.5ml甲醇溶解。更优选地,每15g所述待测中药组合物样品对应的残渣用0.5ml~1.5ml甲醇溶解。
113.优选地,制备甘草对照药材溶液的方法包括:取甘草对照药材,参照上述制备供试品溶液的方法,制备甘草对照药材溶液。更优选地,每0.1g~1g甘草对照药材对应加入水50ml~150ml。加入水饱和的正丁醇30ml~50ml。用水洗脱时,每0.1g~1g甘草对照药材对应加入水10ml~30ml。用甲醇洗脱时,每0.1g~1g甘草对照药材对应加入甲醇5ml~15ml。
114.优选地,制备芍药苷对照品溶液的溶剂为甲醇。更优选地,对照品溶液中,芍药苷对照品的浓度为0.1mg/ml~1mg/ml。
115.优选地,以体积比为(14~16):(0.5~1.5):(0.5~1.5):(1~3)的乙酸乙酯、甲酸、冰醋酸和水为展开剂。
116.优选地,晾干后,还包括喷以体积分数为5%~15%的硫酸的乙醇溶液,加热至斑点显色清晰的步骤
117.可选地,采用高效液相色谱法检测所述待测中药组合物样品中芍药苷的含量的方法包括以下步骤:
118.制备芍药苷对照品溶液;
119.混合待测中药组合物样品和提取溶剂,超声提取,取上清液,通过聚酰胺柱,用水洗脱,收集洗脱液,制备待测中药组合物样品溶液;
120.对所述芍药苷对照品溶液和待测中药组合物样品溶液进行高效液相色谱法检测,根据所得色谱图,计算芍药苷的含量;
121.所述高效液相色谱法检测的色谱条件包括:流动相包括体积比为(13~17):(83~87)的乙腈和磷酸二氢钾溶液,等度洗脱。
122.优选地,所述提取溶剂为水。
123.优选地,每1g~5g待测中药组合物样品对应提取溶剂的用量为10ml~50ml。
124.优选地,超声提取的时间20min~40min
125.优选地,磷酸二氢钾溶液中,磷酸二氢钾的浓度为0.01mol/l~0.1mol/l。
126.优选地,所述高效液相色谱法检测的色谱条件还包括:以十八烷基硅烷键合硅胶为填充剂。
127.优选地,所述高效液相色谱法检测的色谱条件还包括:柱温25℃~40℃。
128.优选地,所述高效液相色谱法检测的色谱条件还包括:检测波长为210~250nm。
129.优选地,所述高效液相色谱法检测的色谱条件还包括:理论板数按芍药苷峰计算应不低于3000。更优选不低于4000。
130.优选地,所述芍药苷对照品溶液的溶剂为水。更优选地,芍药苷对照品溶液中,芍药苷对照品的浓度为0.01mg/ml~0.1mg/ml。
131.可选地,采用高效液相色谱法检测所述待测中药组合物样品中丹酚酸b的含量的方法包括以下步骤:
132.制备丹酚酸b对照品溶液;
133.混合待测中药组合物样品和提取溶剂,超声提取,取提取液,滤过,制备待测中药组合物样品溶液;
134.对所述丹酚酸b对照品溶液和待测中药组合物样品溶液进行高效液相色谱法检测,根据所得色谱图,计算丹酚酸b的含量;
135.所述高效液相色谱法检测的色谱条件包括:流动相包括体积比为(18~25):(75~82)的乙腈和甲酸溶液,等度洗脱。
136.优选地,所述提取溶剂为甲醇溶液。更优选地,甲醇溶液中甲醇的体积分数为70%~80%。
137.优选地,每0.1g~1g待测中药组合物样品对应提取溶剂的用量为10ml~50ml。
138.优选地,超声提取的时间20min~40min
139.优选地,流动相中,甲酸溶液中甲酸的体积分数为1%~2%。
140.优选地,所述高效液相色谱法检测的色谱条件还包括:以十八烷基硅烷键合硅胶为填充剂。
141.优选地,所述高效液相色谱法检测的色谱条件还包括:柱温25℃~40℃。
142.优选地,所述高效液相色谱法检测的色谱条件还包括:检测波长为260~300nm。
143.优选地,所述高效液相色谱法检测的色谱条件还包括:理论板数按丹酚酸b峰计算应不低于3000。更优选不低于4000。
144.优选地,丹酚酸b对照品溶液的溶剂为甲醇溶液。更优选地,甲醇溶液中甲醇的体积分数为70%~80%。更优选地,丹酚酸b对照品溶液中,丹酚酸b对照品的浓度为0.05mg/ml~0.5mg/ml。
145.可选地,采用高效液相色谱法检测所述待测中药组合物样品中黄芪甲苷的含量的方法包括以下步骤:
146.制备黄芪甲苷对照品溶液;
147.混合待测中药组合物样品和提取溶剂,加热回流提取,取提取液,干燥,残渣加水溶解,加水饱和的正丁醇振摇提取,取正丁醇相,碱洗,弃去碱液,干燥,残渣加所述提取溶剂,制备待测中药组合物样品溶液;
148.对所述黄芪甲苷对照品溶液和待测中药组合物样品溶液进行高效液相色谱法检测,根据所得色谱图,计算黄芪甲苷的含量;
149.所述高效液相色谱法检测的色谱条件包括:流动相包括体积比为(25~35):(65~75)的乙腈和水,等度洗脱。
150.优选地,所述提取溶剂为甲醇。
151.优选地,每5g~20g待测中药组合物样品对应加入提取溶剂的用量为50ml~200ml。更优选地,每5g~20g待测中药组合物样品对应加入提取溶剂的用量为95ml~105ml。
152.优选地,加热回流提取的时间3h~5h。
153.优选地,加水饱和的正丁醇振摇提取的次数为3~5次。
154.优选地,每次振摇提取时,每5g~20g待测中药组合物样品对应加入水饱和的正丁醇30ml~50ml。
155.优选地,所述高效液相色谱法检测的色谱条件还包括:以十八烷基硅烷键合硅胶为填充剂。
156.优选地,所述高效液相色谱法检测的色谱条件还包括:蒸发光散射检测器,理论板数按黄芪甲苷峰计算应不低于3000。更优选不低于4000。
157.更优选地,设置蒸发光散射检测器参数如下:漂移管温度105℃,载气流量:2.8l/min;柱温25℃~40℃。
158.优选地,黄芪甲苷对照品溶液的溶剂为甲醇。更优选地,黄芪甲苷对照品溶液中,黄芪甲苷对照品的浓度为0.05mg/ml~0.5mg/ml。
159.可选地,采用高效液相色谱法检测所述待测中药组合物样品中2,3,5,4
’‑
四羟基二苯乙烯-2-o-β-d葡萄糖苷的含量的方法包括以下步骤:
160.制备2,3,5,4
’‑
四羟基二苯乙烯-2-o-β-d葡萄糖苷对照品溶液;
161.混合待测中药组合物样品和提取溶剂,超声提取,取提取液,滤过,制备待测中药组合物样品溶液;
162.对所述2,3,5,4
’‑
四羟基二苯乙烯-2-o-β-d葡萄糖苷对照品溶液和待测中药组合物样品溶液进行高效液相色谱法检测,根据所得色谱图,计算2,3,5,4
’‑
四羟基二苯乙烯-2-o-β-d葡萄糖苷的含量;
163.所述高效液相色谱法检测的色谱条件包括:流动相包括体积比为(13~18):(82~87)的乙腈和水作为流动相,等度洗脱。
164.优选地,所述提取溶剂为乙醇溶液。更优选地,乙醇溶液中乙醇的体积分数为20%~50%。
165.优选地,每3g~8g待测中药组合物样品对应提取溶剂的用量为25ml~100ml。
166.优选地,超声提取的时间20min~40min。
167.优选地,所述高效液相色谱法检测的色谱条件还包括:以十八烷基硅烷键合硅胶为填充剂。
168.优选地,所述高效液相色谱法检测的色谱条件还包括:柱温25℃~40℃。
169.优选地,所述高效液相色谱法检测的色谱条件还包括:检测波长为310~330nm。
170.优选地,所述高效液相色谱法检测的色谱条件还包括:理论板数按2,3,5,4
’‑
四羟基二苯乙烯-2-o-β-d葡萄糖苷峰计算应不低于3000。更优选不低于4000。
171.优选地,2,3,5,4
’‑
四羟基二苯乙烯-2-o-β-d葡萄糖苷峰对照品溶液的溶剂为乙醇溶液。更优选地,乙醇溶液中乙醇的体积分数为20%~50%。更优选地,2,3,5,4
’‑
四羟基二苯乙烯-2-o-β-d葡萄糖苷峰对照品溶液中,2,3,5,4
’‑
四羟基二苯乙烯-2-o-β-d葡萄糖苷峰对照品的浓度为0.5mg/ml~2.5mg/ml。
172.可选地,采用高效液相色谱法检测所述待测中药组合物样品中苦参碱的含量的方法包括以下步骤:
173.制备苦参碱对照品溶液;
174.混合待测中药组合物样品、水和氨水,超声至溶解,加三氯甲烷溶液萃取,取三氯甲烷相,去除溶剂,残渣加流动相溶解,滤过,制备待测中药组合物样品溶液;
175.对所述苦参碱对照品溶液和待测中药组合物样品溶液进行高效液相色谱法检测,根据所得色谱图,计算苦参碱的含量;
176.所述高效液相色谱法检测的色谱条件包括:流动相包括乙腈、磷酸溶液和三乙胺,其中,乙腈和磷酸溶液的体积比为(18~22):(78~82),三乙胺调节所述流动向的ph值为8.0
±
0.1,等度洗脱。
177.优选地,流动相中,磷酸溶液中磷酸的体积分数为0.05%~0.15%。
178.优选地,每0.5g~5g待测中药组合物样品对应加入水的用量为10ml~50ml。
179.优选地,每0.5g~5g待测中药组合物样品对应加入氨水的用量为0.1ml~0.5ml。
180.优选地,加三氯甲烷溶液萃取的次数为2~5次,合并三氯甲烷相。
181.优选地,每次萃取时,每0.5g~5g待测中药组合物样品对应加入三氯甲烷的用量为10ml~20ml。
182.优选地,所述高效液相色谱法检测的色谱条件还包括:以十八烷基硅烷键合硅胶为填充剂。
183.优选地,所述高效液相色谱法检测的色谱条件还包括:检测波长为210~230nm。
184.优选地,所述高效液相色谱法检测的色谱条件还包括:柱温为25~30℃。
185.优选地,所述高效液相色谱法检测的色谱条件还包括:理论板数按苦参碱峰计算应不低于4000。
186.优选地,苦参碱对照品溶液的溶剂为流动相。更优选地,流动相中,磷酸溶液中磷酸的体积分数为0.05%~0.15%。更优选地,苦参碱对照品溶液中,苦参碱对照品的浓度为0.05mg/ml~0.5mg/ml。
187.可选地,所述中药组合物的理论制备原料中,大黄、黄芪、桑白皮、苦参、党参、白术、茯苓、制何首乌、白芍、丹参、川芎、菊花、姜半夏、车前草、柴胡和甘草的重量比为(0.5~1.5):(3-5):(2-4):(1-3):(2-4):(4-6):(4-6):(4-6):(2-4):(4-6):(2-4):(1.5-3.5):
(1-3):(4-6):(1-2):(0.5-1.5)。
188.优选地,所述中药组合物的理论制备原料中,大黄、黄芪、桑白皮、苦参、党参、白术、茯苓、制何首乌、白芍、丹参、川芎、菊花、姜半夏、车前草、柴胡和甘草的重量比为1:4:3:2:3:5:5:5:3:5:3:2.5:2:5:1.5:0.9。
189.可选地,所述中药组合物的理论制备方法包括以下步骤:
190.向所述大黄、黄芪、桑白皮、苦参、党参、白术、茯苓、制何首乌、白芍、丹参、川芎、菊花、姜半夏、车前草、柴胡和甘草的混合物中加水提取,收集滤液,加入药用辅料,制粒。
191.相比于传统方案,本发明有益效果如下:
192.本发明经过多大量研究建立针对特定中药组合物的质量检测方法。检测方法专属性强,重现性、稳定性及精密度良好,能够有效控制中药组合物的质量,使本发明的中药组合物的质量达到稳定、可控、高效及安全,有利于对其进行全面质量控制。
附图说明
193.图1为大黄和制何首乌tlc鉴别结果图谱;
194.图2为黄芪tlc鉴别结果图谱;
195.图3为丹参tlc鉴别结果图谱;
196.图4为苦参tlc鉴别结果图谱;
197.图5为柴胡tlc鉴别结果图谱;
198.图6为桑白皮tlc鉴别结果图谱;
199.图7为茯苓tlc鉴别结果图谱;
200.图8为党参tlc鉴别结果图谱;
201.图9为甘草和白芍tlc鉴别结果图谱。
具体实施方式
202.以下结合具体实施例对本发明作进一步详细的说明。本发明可以以许多不同的形式来实现,并不限于本文所描述的实施方式。相反地,提供这些实施方式的目的是使对本发明公开内容理解更加透彻全面。
203.除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不是旨在于限制本发明。
204.若无特殊说明,本发明具体实施例所用试剂、仪器均为市售普通产品。
205.若无特殊说明,本发明所述“溶液”的溶剂为水。例如硫酸溶液为硫酸的水溶液,氢氧化钠溶液为氢氧化钠的水溶液。
206.1试药
207.本发明的中药组合物为尿毒清颗粒,由康臣药业(内蒙古)责任有限公司提供,其制备方法为:
208.1)按大黄、黄芪、桑白皮、苦参、党参、白术、茯苓、制何首乌、白芍、丹参、川芎、菊花、姜半夏、车前草、柴胡和甘草的重量比为1:4:3:2:3:5:5:5:3:5:3:2.5:2:5:1.5:0.9准备以上药材。
209.2)向以上十六味药材的混合物中加水提取两次,滤过,合并滤液,浓缩至流浸膏,收膏备用;
210.3)取以上流浸膏,加适量药用辅料,喷雾制粒得颗粒剂;干燥,过筛取合格颗粒,质检,包装,即得颗粒成品。
211.2薄层色谱法检测
212.2.1仪器、试剂
213.仪器:研钵、量筒、圆底烧瓶、冷凝管、蒸发皿、层析缸等均购于广州市东征化玻仪器有限公司、电热套(巩义市予华仪器有限责任公司)、bs224s分析天平(北京赛多利斯仪器系统有限公司)、linomat5半自动点样仪(瑞士卡玛)、硅胶g薄层板(青岛海洋化工厂有限公司)、reprostar3成像系统(瑞士卡玛)、gzx-gfc-01-2-bs烘箱(上海博泰实验设备有限公司)、th-ii加热器(上海科哲生化科技有限公司)。
214.试剂:甲醇、盐酸、乙酸乙酯、三氯甲烷、水、石油醚(60~90℃沸程)、氢氧化钠、硫酸、乙酸、正丁醇、氯仿、硫酸乙醇试液。
215.2.2对照药材、对照品
216.对照药材:大黄对照药材、制何首乌对照药材、丹参对照药材、柴胡对照药材、桑白皮对照药材、茯苓对照药材、党参对照药材、甘草对照药材,购于中国食品药品检定研究院。
217.对照品:大黄酸、大黄素、大黄素甲醚、黄芪甲苷、原儿茶醛、苦参碱、党参炔苷、芍药苷,购于中国食品药品检定研究院。
218.2.3大黄和制何首乌的鉴别
219.2.3.1供试品溶液的制备
220.取“1”项下的中药组合物5g,加入3mol/l硫酸溶液30ml,加热回流1小时,放冷;再加入氯仿(三氯甲烷)30ml,加热回流1小时,放冷,分取氯仿(三氯甲烷)液,蒸干,残渣加1ml氯仿(三氯甲烷)溶解,作为供试品溶液。
221.2.3.2对照药材溶液和对照品混合溶液的制备
222.取大黄对照药材和制何首乌对照药材各1g,分别加入3mol/l硫酸溶液30ml,加热回流1小时,放冷;再加入氯仿(三氯甲烷)30ml,加热回流1小时,放冷,分取氯仿(三氯甲烷)液,蒸干,残渣加1ml氯仿(三氯甲烷)溶解,制得大黄对照药材溶液和制何首乌对照药材溶液。
223.取大黄酸、大黄素和大黄素甲醚对照品适量,加入甲醇,制成含1mg/ml大黄酸对照品、1mg/ml大黄素对照品和1mg/ml大黄素甲醚对照品的混合溶液,作为对照品混合溶液。
224.2.3.3照薄层色谱法(《中国药典》2020版四部通则)试验,分别吸取所述供试品溶液、大黄对照药材溶液、制何首乌对照药材溶液以及对照品混合溶液,点于同一硅胶h薄层板上,以体积比为15:5:1的石油醚(沸点为30℃~60℃)、甲酸乙酯和甲酸的上层溶液为展开剂,展开,取出晾干,置于紫外光或氨蒸气熏后置日光下观察,分别显橙色荧光斑点和红色斑点。结果见图1,图1中的1~6为供试品溶液;7为大黄对照药材溶液;8为制何首乌对照药材溶液;9为对照品混合溶液,其中,a为置uv365nm检视,b为置可见光下检视。
225.2.4黄芪的鉴别
226.2.4.1供试品溶液的制备
227.取“1”项下的中药组合物5g,加正丁醇20ml,加热回流2小时,放冷,滤过,滤液用质
量分数为1%氢氧化钠溶液洗涤3次,每次15ml,弃去碱液,再用水饱和的正丁醇洗至中性,弃去水层,正丁醇提取液置水浴上蒸干,残渣加甲醇0.2ml溶解,作为供试品溶液。
228.2.4.2对照品溶液的制备
229.取黄芪甲苷对照品适量,加甲醇制成1mg/ml的溶液,作为对照品溶液。
230.2.4.3照薄层色谱法(《中国药典》2020版四部通则)试验,分别吸取所述供试品溶液和对照品溶液,点于同一硅胶g薄层板上,以体积比为13:7:2的氯仿(三氯甲烷)、甲醇和水的下层溶液为展开剂,展开,取出晾干,喷以质量分数为10%的硫酸的乙醇溶液,加热至斑点显色清晰,分别置可见光下以及紫外光灯下检视。结果见图2,图2中的1~6为供试品溶液;7为黄芪甲苷对照品溶液,其中,a为置uv365nm检视,b为置可见光下检视。
231.2.5丹参的鉴别
232.2.5.1供试品溶液的制备
233.取“1”项下的中药组合物5g,加水50ml溶解,滤过,滤液用乙醚提取2次,每次15ml,合并乙醚相,蒸干,残渣加2ml乙醇溶解,作为供试品溶液。
234.2.5.2对照药材溶液和对照品溶液的制备
235.取丹参对照药材5g,加水煎煮2次,每次10分钟,合并煎液,浓缩至10ml流浸膏,取5ml浸膏,加水50ml溶解,滤过,滤液用乙醚提取2次,每次15ml,合并乙醚相,蒸干,残渣加2ml乙醇溶解,制得对照药材溶液。
236.取原儿茶醛对照品适量,加入乙醇制备成0.3mg/ml溶液,作为对照品溶液。
237.2.5.3照薄层色谱法(《中国药典》2020版四部通则)试验,分别吸取所述供试品溶液、对照药材溶液以及对照品溶液,点于同一硅胶g薄层板上,以体积比为2:4:4:0.5的己烷、苯、乙酸乙酯和甲酸作为展开剂,展开,取出晾干,喷以质量分数为0.1%的2、4-二硝基苯肼的乙醇溶液,置于日光下观察。结果见图3,图3中的1~6为供试品溶液;7为原儿茶醛对照品溶液;8为丹参对照药材溶液。
238.2.6苦参的鉴别
239.2.6.1供试品溶液的制备
240.取“1”项下的中药组合物5g,加氯仿(三氯甲烷)50ml、浓氨溶液0.3ml,混均,放置过夜,滤过,滤液蒸干,残渣加氯仿(三氯甲烷)0.2ml溶解,作为供试品溶液。
241.2.6.2对照品溶液的制备
242.取苦参碱对照品适量,加乙醇制成0.2mg/ml溶液,作为对照品溶液。
243.2.6.3照薄层色谱法(《中国药典》2020版四部通则)试验,分别吸取所述供试品溶液和对照品溶液,点于同一硅胶g薄层板上,以体积比为20:20:3:1的甲苯、乙酸乙酯、甲醇和浓氨溶液为展开剂,展开,取出晾干,喷以稀碘化铋试液,置可见光下检视。结果见图4,图4中的1~6为供试品溶液;7为苦参碱对照品溶液。
244.2.7柴胡的鉴别
245.2.7.1供试品溶液的制备
246.取“1”项下的中药组合物5g,加水30ml,超声提取20分钟,放冷,滤过,滤液通过ab-8型大孔吸附树脂柱(内径为1.5cm,柱高为10cm),用体积分数为50%的乙醇溶液50ml洗脱,弃去洗脱液,再用体积分数为90%的乙醇溶液50ml洗脱,收集洗脱液,水浴蒸干,残渣加1ml甲醇溶解,作为供试品溶液。
247.2.7.2对照药材溶液的制备
248.取柴胡对照药材0.2g,加甲醇适量,超声提取,滤过,滤液浓缩至适量,作为对照药材溶液。
249.2.7.3照薄层色谱法(《中国药典》2020版四部通则)试验,分别吸取所述供试品溶液和对照药材溶液,点于同一硅胶g薄层板上,以体积比为11:2:1的乙酸乙酯、乙醇和水为展开剂,展开,取出晾干,喷以质量分数为2%的对二甲基甲醛的硫酸水溶液,其中硫酸水溶液中硫酸的质量分数为40%,加热至斑点显色清晰,分别置可见光下以及紫外光灯下检视。结果见图5,图5中的1~3为供试品溶液;4为柴胡对照药材溶液。其中,a为置uv365nm检视,b为置可见光下检视。
250.2.8桑白皮的鉴别
251.2.8.1供试品溶液的制备
252.取“1”项下的中药组合物2g,加饱和碳酸钠溶液20ml,超声提取20分钟,放冷,滤过,滤液加稀盐酸调节ph值至1~2,静置30分钟,滤过,滤液用乙酸乙酯振摇提取2次,每次20ml,合并乙酸乙酯相,水浴蒸干,残渣加1ml甲醇溶解,作为供试品溶液。
253.2.8.2对照药材溶液
254.取桑白皮对照药材2g,加饱和碳酸钠溶液20ml,超声提取20分钟,放冷,滤过,滤液加稀盐酸调节ph值至1~2,静置30分钟,滤过,滤液用乙酸乙酯振摇提取2次,每次20ml,合并乙酸乙酯相,水浴蒸干,残渣加1ml甲醇溶解,制得对照药材溶液。
255.2.8.3照薄层色谱法(《中国药典》2020版四部通则)试验,分别吸取所述供试品溶液和对照药材溶液,点于同一聚酰胺薄层板上,以醋酸为展开剂,展开,取出晾干,置紫外光灯下检视。结果见图6,图6中的1~3为供试品溶液;4~5为桑白皮对照药材溶液。
256.2.9茯苓的鉴别
257.2.9.1供试品溶液的制备
258.取“1”项下的中药组合物5g,加水40ml,超声提取30分钟,放冷,滤过,滤液用水饱和的正丁醇萃取3次,每次20ml,合并正丁醇相,蒸干,残渣加2ml甲醇溶解,作为供试品溶液。
259.2.9.2对照药材溶液
260.取茯苓对照药材1g,加水40ml,超声提取30分钟,放冷,滤过,滤液用水饱和的正丁醇萃取3次,每次20ml,合并正丁醇相,蒸干,残渣加2ml甲醇溶解,制得对照药材溶液。
261.2.9.3照薄层色谱法(《中国药典》2020版四部通则)试验,分别吸取所述供试品溶液和对照药材溶液,点于同一硅胶g薄层板上,以体积比20:5:0.5的甲苯、乙酸乙酯和甲酸为展开剂,展开,取出晾干,喷以质量分数为2%的香草醛的硫酸溶液-乙醇(4:1,v/v)混合溶液,加热至斑点显色清晰,置日光下检视。分别置可见光下以及紫外光灯下检视。结果见图7,图7中的1~3为供试品溶液;4~5为茯苓对照药材溶液。其中,a为置uv365nm检视,b为置可见光下检视。
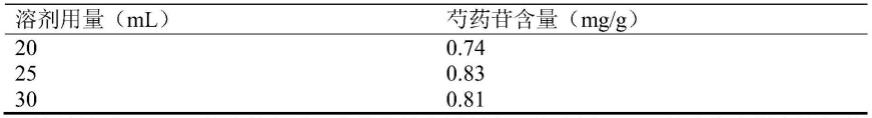
262.2.10党参的鉴别
263.2.10.1供试品溶液的制备
264.取“1”项下的中药组合物5g,加水30ml,超声提取20分钟,放冷,滤过,滤液通过ab-8型大孔吸附树脂柱(内径为1.5cm,柱高为10cm),用体积分数为50%的乙醇溶液50ml洗脱,
弃去洗脱液,再用体积分数为90%的乙醇溶液50ml洗脱,收集洗脱液,水浴蒸干,残渣加甲醇溶解,作为供试品溶液。
265.2.10.2对照药材溶液和对照品混合溶液的制备
266.取党参对照药材1g,加水30ml,超声提取20分钟,放冷,滤过,滤液通过ab-8型大孔吸附树脂柱(内径为1.5cm,柱高为10cm),用50%乙醇50ml洗脱,弃去洗脱液,再用90%乙醇50ml洗脱,收集洗脱液,水浴蒸干,残渣加甲醇溶解,制得对照药材溶液。
267.取党参炔苷对照品适量,加甲醇制成1mg/ml溶液,作为对照品溶液。
268.2.10.3照薄层色谱法(《中国药典》2020版四部通则)试验,分别吸取所述供试品溶液、对照药材溶液和对照品溶液,点于同一硅胶g薄层板上,以体积比为7:1:0.5的正丁醇、冰醋酸和水为展开剂,展开,取出晾干,喷以质量分数为10%的硫酸的乙醇溶液,加热至斑点显色清晰,分别置可见光下以及紫外光灯下检视。结果见图8,图8中的1~3为供试品溶液;4为党参对照药材溶液;5为党参炔苷对照品溶液。
269.2.11甘草和白芍的鉴别
270.2.11.1供试品溶液的制备
271.取“1”项下的中药组合物15g,加水100ml溶解,加水饱和的正丁醇萃取2次,每次40ml,合并正丁醇相,蒸干,残渣加水3ml溶解,加于已处理好的c
18
小柱(先以少量甲醇浸渍,再以水洗去甲醇备用)上,分别用水20ml、甲醇10ml洗脱,收集甲醇洗脱液后蒸干,残渣用1ml甲醇溶解,作为供试品溶液。
272.2.11.2对照药材溶液和对照品混合溶液的制备
273.取甘草对照药材0.5g,加水100ml溶解,加水饱和的正丁醇萃取2次,每次40ml,合并正丁醇相,蒸干,残渣加水3ml溶解,加于已处理好的c
18
小柱(先以少量甲醇浸渍,再以水洗去甲醇备用)上,分别用水20ml、甲醇10ml洗脱,收集甲醇洗脱液后蒸干,残渣用1ml甲醇溶解,制得对照药材溶液。
274.取芍药苷对照品适量,加甲醇制成0.5mg/ml溶液,作为对照品溶液。
275.2.11.3照薄层色谱法(《中国药典》2020版四部通则)试验,分别吸取所述供试品溶液、对照药材和对照品溶液,点于同一含碱性硅胶g层板(硅胶g预制板浸以0.3%naoh乙醇溶液)上,以体积比为15:1:1:2的乙酸乙酯、甲酸、冰醋酸和水为展开剂,展开,取出晾干,喷以体积分数为10%的硫酸的乙醇溶液,加热至斑点显色清晰,分别置可见光下以及紫外光灯下检视。结果见图9,图9中的1~10为供试品溶液;11为芍药苷对照品溶液;12为甘草对照药材溶液,a为置uv365nm检视,b为置可见光下检视。
276.3高效液相色谱法检测
277.3.1芍药苷含量测定
278.3.1.1仪器、试剂
279.仪器:agilent1260高效液相色谱仪;dad检测器;色谱柱:phenomenex c
18
(250
×
4.6mm,5μm);kq3200db型数控超声仪(巩义市予华仪器有限责任公司);电热套(巩义市予华仪器有限责任公司)、bs224s分析天平(北京赛多利斯仪器系统有限公司);研钵、量筒、圆底烧瓶、冷凝管、蒸发皿等均购于广州市东征化玻仪器有限公司;电热套(巩义市予华仪器有限责任公司)。
280.试药:乙腈(色谱纯);超纯水;甲酸(色谱纯),乙醇、甲醇、芍药苷对照品、待测中药
组合物样品(批号:20140801,20140802,20140803,均由康臣药业(内蒙古)责任有限公司提供)。
281.3.1.2对照品溶液、供试品溶液、阴性样品溶液的制备
282.对照品溶液:80℃干燥至恒重的芍药苷对照品10mg,精密称定,置50ml量瓶中,加水溶解定容至刻度,精密量取5ml,置25ml量瓶中,加水至刻度,摇匀,即得对照品溶液。
283.供试品溶液:取“1”项下的中药组合物3g,精密称定,置具塞锥形瓶中,精密加水25ml,摇匀,超声处理30min(功率120w,频率40khz),放冷,再称重,用水补足减少的重量,摇匀,滤过,精密吸收上清液5ml,加于聚酰胺柱(30~60目,1cm
×
20cm)上,用水洗脱,准确收集洗脱液25ml,摇匀,即可。
284.阴性样品溶液:取不含白芍的处方量药材,按“1”项下的中药组合物的制备工艺及“3.1.2”项下的供试品溶液制备方法制备阴性样品溶液。
285.3.1.3色谱条件
286.色谱柱:phenomenex c
18
(250
×
4.6mm,5μm)
287.流动相:乙腈:0.05mol/l的磷酸二氢钾溶液=15:85(v/v),等度洗脱
288.检测波长:230nm
289.流速:1.0ml/min
290.柱温:25℃
291.进样量:10μl
292.3.1.4供试品溶液制备方法的考察
293.3.1.4.1溶剂用量的考察
294.取“1”项下的中药组合物3g,精密称定,除了水的用量分别为20ml、25ml、30ml外,其他条件按“3.1.2”项下的供试品溶液制备方法进行,制备供试品溶液。将所得供试品溶液和“3.1.2”项下的对照品溶液分别按“3.1.3”项下的色谱条件进样至高效液相色谱仪中测定,根据所得色谱图计算芍药苷的含量,结果如表1所示。
295.表1
[0296][0297]
结果显示:25ml的水提取时,芍药苷的含量最高,故选用25ml的水作为提取溶剂的用量。
[0298]
3.1.4.2超声时间的考察
[0299]
取“1”项下的中药组合物3g,精密称定,除了超声时间分别为20min、30min、40min外,其他条件按“3.1.2”项下的供试品溶液制备方法进行,制备供试品溶液。将所得供试品溶液和“3.1.2”项下的对照品溶液分别按“3.1.3”项下的色谱条件进样至高效液相色谱仪中测定,根据所得色谱图计算芍药苷的含量,结果如表2所示。
[0300]
表2
[0301][0302]
结果显示:超声提取30min时,芍药苷的含量最高,故选用30min作为超声提取的时间。
[0303]
3.1.4.3供试品溶液的制备方法的确认
[0304]
取本品3g,精密称定,置具塞锥形瓶中,精密加水25ml,摇匀,超声处理30min(功率120w,频率40khz),放冷,再称重,用水补足减少的重量,摇匀,滤过,精密吸收上清液5ml,加于聚酰胺柱(30~60目,1cm
×
20cm)上,用水洗脱,准确收集洗脱液25ml,摇匀,即得供试品溶液。
[0305]
3.1.5色谱条件的考察
[0306]
3.1.5.1波长的确定
[0307]
根据《中国药典》2020版一部芍药苷含量测定方法,确定芍药苷的检测波长为230nm。
[0308]
3.1.5.2色谱柱的考察
[0309]
取“1”项下的中药组合物3g,精密称定,按“3.1.4.3”项下的供试品溶液制备方法制备供试品溶液,将所得供试品溶液进样至高效液相色谱仪中测定,其中,除了色谱柱分别为phenomenex、agilent、alltima(规格见表3)外,其他条件按“3.1.3”项下的色谱条件进行,根据所得色谱图观察分离度和理论塔板数,结果如表3所示。
[0310]
表3
[0311][0312]
结果显示:phenomenex色谱柱分离效果最好。
[0313]
3.1.5.3流动相的考察
[0314]
取“1”项下的中药组合物3g,精密称定,按“3.1.4.3”项下的供试品溶液制备方法制备供试品溶液,将所得供试品溶液进样至高效液相色谱仪中测定,其中,除了流动相分别为体积比为15:85的乙腈-0.05mol/l的磷酸二氢钾溶液、体积比为14:86的乙腈-0.05mol/l的磷酸二氢钾溶液、体积比为16:84的乙腈-0.05mol/l的磷酸二氢钾溶液外,其他条件按“3.1.3”项下的色谱条件进行,根据所得色谱图观察分离度。
[0315]
结果显示:体积比为15:85的乙腈-0.05mol/l的磷酸二氢钾溶液的分离度最好,确定其为流动相。
[0316]
3.1.5.4柱温的考察
[0317]
取“1”项下的中药组合物3g,精密称定,按“3.1.4.3”项下的供试品溶液制备方法制备供试品溶液,将所得供试品溶液进样至高效液相色谱仪中测定,其中,除了柱温分别为25℃、30℃、35℃外,其他条件按“3.1.3”项下的色谱条件进行,根据所得色谱图观察分离度。
[0318]
结果显示:柱温对分离度无影响,本方法选择柱温为25℃。
[0319]
3.1.5.5色谱条件的确认
[0320]
色谱柱:phenomenex c
18
(250
×
4.6mm,5μm)
[0321]
流动相:乙腈:0.05mol/l的磷酸二氢钾溶液=15:85(v/v),等度洗脱
[0322]
检测波长:230nm
[0323]
流速:1.0ml/min
[0324]
柱温:25℃
[0325]
进样量:10μl
[0326]
3.1.6系统适应性研究和方法学验证
[0327]
3.1.6.1系统适应性研究
[0328]
色谱分离:在“3.1.5.5”项下,芍药苷的色谱峰保留时间约为12min,与其它峰分离良好,分离度大于1.5,对称性为0.98,符合标准。
[0329]
理论塔板数:按公式n=5.54(tr/w
h/2
)计算芍药苷的理论塔板数为13744,考虑到不同色谱柱条件:柱长、载体性能、填充情况、流动相、使用时间等差异,暂定芍药苷色谱峰的理论塔板数不小于4000。
[0330]
3.1.6.2专属性试验
[0331]
分别取提取溶剂(空白水溶剂)、“3.1.2”项下的对照品溶液,“3.1.4.3”项下的供试品溶液以及“3.1.2”项下的阴性样品溶液各10μl,按“3.1.5.5”项下的色谱条件进样至高效液相色谱仪中测定。
[0332]
结果显示:阴性无干扰。
[0333]
3.1.6.3线性范围考察
[0334]
取芍药苷对照品,加水制备芍药苷对照品储备液适量,精密量取芍药苷对照品储备液,倍比稀释成浓度为101.00、50.500、25.250、12.625、6.313μg/ml的对照品溶液。将所得各浓度的对照品溶液按“3.1.5.5”项下的色谱条件进样至高效液相色谱仪中测定,根据所得色谱图,记录峰面积,以浓度为横坐标,峰面积(a1、a2、a3)为纵坐标,绘制标准曲线,结果如表4所示,回归方程为:y=22.549x+56.772(r2=0.9999)。
[0335]
表4
[0336][0337]
结果显示,在0.06313~1.01000μg范围内线性关系良好。
[0338]
3.1.6.4精密度试验
[0339]
取浓度为40.400μg/ml芍药苷对照品溶液,按“3.1.5.5”项下的色谱条件,连续进样至高效液相色谱仪中测定5次,根据所得色谱图,记录峰面积,计算芍药苷的含量,计算rsd,结果如表5所示。
[0340]
表5
[0341][0342]
结果表明,本方法精密度良好。
[0343]
3.1.6.5稳定性试验
[0344]
取“1”项下的中药组合物3g,精密称定,按“3.1.4.3”项下的供试品溶液的制备方法制备供试品溶液,按“3.1.5.5”项下的色谱条件,在0、2、4、8、24、48、96小时进样至高效液相色谱仪中测定,根据所得色谱图,记录峰面积,计算芍药苷的含量,计算rsd,结果如表6所示。
[0345]
表6
[0346][0347]
结果显示,本方法96小时内稳定。
[0348]
3.1.6.6重复性试验
[0349]
取“1”项下的中药组合物3g,精密称定,按“3.1.4.3”项下的供试品溶液的制备方法制备6份供试品溶液,按“3.1.5.5”项下的色谱条件进样至高效液相色谱仪中测定,根据所得色谱图,记录峰面积,计算芍药苷的含量,计算rsd,结果如表7所示。
[0350]
表7
[0351][0352]
结果显示,本方法重复性良好。
[0353]
3.1.6.7加样回收试验
[0354]
取“1”项下的中药组合物1.5g,平行6份,精密称定,分别精密加入浓度为0.4040mg/ml的芍药苷对照品1ml,挥干,按“3.1.4.3”项下的供试品溶液的制备方法制备6份供试品溶液,按“3.1.5.5”项下的色谱条件进样至高效液相色谱仪中测定,根据所得色谱图计算芍药苷的含量、回收率及rsd值,结果如表8所示。
[0355]
表8
[0356][0357][0358]
3.1.7三批样品含量测定
[0359]
取3批(批号见表9)待测中药组合物样品,3g,精密称定,平行2份,按“3.1.4.3”项下的供试品溶液的制备方法制备待测中药组合物样品溶液,将所得待测中药组合物样品溶液和“3.1.2”项下的对照品溶液分别按“3.1.5.5”项下的色谱条件进样至高效液相色谱仪中测定,根据所得色谱图计算芍药苷的含量,结果如表9所示。
[0360]
表9
[0361][0362]
结果显示,三批样品中芍药苷的含量均大于0.7mg/g。
[0363]
3.2丹酚酸b含量测定
[0364]
3.2.1仪器、试剂
[0365]
仪器:agilent1260高效液相色谱仪;dad检测器;色谱柱:agilent extend-c
18
(250
×
4.6mm,5μm);kq3200db型数控超声仪(巩义市予华仪器有限责任公司);电热套(巩义市予华仪器有限责任公司)、bs224s分析天平(北京赛多利斯仪器系统有限公司);研钵、量筒、圆底烧瓶、冷凝管、蒸发皿等均购于广州市东征化玻仪器有限公司;电热套(巩义市予华仪器有限责任公司)。
[0366]
试药:乙腈(色谱纯);超纯水;甲酸(色谱纯),乙醇、甲醇、丹酚酸b对照品、待测中药组合物样品(批号:20140801,20140802,20140803,均由康臣药业(内蒙古)责任有限公司提供)。
[0367]
3.2.2对照品溶液、供试品溶液、阴性样品溶液的制备
[0368]
对照品溶液:精密称定丹酚酸b对照品10.6mg于25ml容量瓶中,加入体积分数为75%的甲醇溶液定容至刻度,制成浓度为0.424mg/ml的对照品储备溶,取3ml定容至10ml,得浓度为0.1272mg/ml的对照品溶液。
[0369]
供试品溶液:取“1”项下的中药组合物适量,用研钵研碎,过筛,精密称定0.5g于25ml容量瓶中,加入体积分数为75%的甲醇溶液定容至刻度,超声30min,取出放冷,用体积分数为75%的甲醇溶液补足减少量,用0.45μm滤膜滤过,即得供试品溶液。
[0370]
阴性样品溶液:取不含丹参的处方量药材,按“1”项下的中药组合物的制备工艺及“3.2.2”项下的供试品溶液制备方法制备阴性样品溶液。
[0371]
3.2.3色谱条件
[0372]
色谱柱:agilent extend-c
18
(250
×
4.6mm,5μm)
[0373]
流动相:乙腈-体积分数为1.5%的甲酸溶液=21:79(v/v),等度洗脱
[0374]
检测波长:286nm
[0375]
流速:1.0ml/min
[0376]
柱温:30℃
[0377]
进样量:10μl
[0378]
3.2.4色谱条件的考察
[0379]
3.2.4.1波长的确定
[0380]
根据《中国药典》2020版一部丹酚酸b含量测定方法,确定丹酚酸b的检测波长为286nm。
[0381]
3.2.4.2色谱柱的考察
[0382]
取“1”项下的中药组合物适量,按“3.2.2”项下的供试品溶液制备方法制备供试品溶液,将所得供试品溶液进样至高效液相色谱仪中测定,其中,除了色谱柱分别为phenomenex、agilent、alltima(规格见表10)外,其他条件按“3.2.3”项下的色谱条件进行,根据所得色谱图观察分离度和理论塔板数,结果如表10所示。
[0383]
表10
[0384][0385]
结果显示:agilent色谱柱分离效果最好。
[0386]
3.2.4.3流动相的考察
[0387]
取“1”项下的中药组合物适量,按“3.2.2”项下的供试品溶液制备方法制备供试品溶液,将所得供试品溶液进样至高效液相色谱仪中测定,其中,除了流动相分别为体积比21:79的乙腈-体积分数为1.5%的甲酸溶液、体积比为20:80的乙腈-体积分数为1.5%的甲酸溶液,体积比为22:78的乙腈-体积分数为1.5%的甲酸溶液外,其他条件按“3.2.3”项下的色谱条件进行,根据所得色谱图观察分离度。
[0388]
结果显示:体积比21:79的乙腈-体积分数为1.5%的甲酸溶液的分离度最好,确定其为流动相。
[0389]
3.2.4.4柱温的考察
[0390]
取“1”项下的中药组合物适量,按“3.2.2”项下的供试品溶液制备方法制备供试品溶液,将所得供试品溶液进样至高效液相色谱仪中测定,其中,除了柱温分别为25℃、30℃、35℃外,其他条件按“3.2.3”项下的色谱条件进行,根据所得色谱图观察分离度。
[0391]
结果显示:柱温对分离度无影响,本方法选择柱温为30℃。
[0392]
3.2.4.5色谱条件的确认
[0393]
色谱柱:agilent extend-c
18
(250
×
4.6mm,5μm)
[0394]
流动相:乙腈-体积分数为1.5%的甲酸溶液=21:79(v/v),等度洗脱
[0395]
检测波长:286nm
[0396]
流速:1.0ml/min
[0397]
柱温:30℃
[0398]
进样量:10μl
[0399]
3.2.5系统适应性研究和方法学验证
[0400]
3.2.5.1系统适应性研究
[0401]
色谱分离:在“3.2.4.5”项下,丹酚酸b的色谱峰保留时间约为16min,与其它峰分离良好,分离度大于1.5,对称性为0.99,符合标准。
[0402]
理论塔板数:按公式n=5.54(tr/w
h/2
)计算丹酚酸b峰的理论塔板数为12557,考虑到不同色谱柱条件:柱长、载体性能、填充情况、流动相、使用时间等差异,暂定丹酚酸b色谱峰的理论塔板数不小于4000。
[0403]
3.2.5.2专属性试验
[0404]
分别取提取溶剂、“3.2.2”项下的对照品溶液,“3.2.2”项下的供试品溶液以及“3.2.2”项下的阴性样品溶液各10μl,按“3.2.4.5”项下的色谱条件进样至高效液相色谱仪中测定。
[0405]
结果表明:阴性无干扰。
[0406]
3.2.5.3线性范围考察
[0407]
精密称取丹酚酸b对照品72.4mg,加甲醇适量溶于50ml的容量瓶中,超声处理1h,浓度为0.144mg/ml。分别吸取5,10,15,20,25,30μl,按“3.2.4.5”项下的色谱条件进样至高效液相色谱仪中测定,根据所得色谱图,记录峰面积,以浓度为横坐标,峰面积为纵坐标,绘制标准曲线,结果如表11所示,回归方程为:y=989225x+7436.1(r2=0.9999)。
[0408]
表11
[0409][0410]
结果显示,在0.72~4.32μg范围内线性关系良好。
[0411]
3.2.5.4精密度试验
[0412]
取浓度为0.1272mg/ml丹酚酸b对照品溶液,按“3.2.4.5”项下的色谱条件,连续进样至高效液相色谱仪中测定5次,根据所得色谱图,记录峰面积,计算丹酚酸b的含量,计算rsd,得到rsd为0.31%。
[0413]
结果显示,本方法精密度良好。
[0414]
3.2.5.5稳定性试验
[0415]
取“1”项下的中药组合物适量,按“3.2.2”项下的供试品溶液的制备方法制备供试品溶液,按“3.2.4.5”项下的色谱条件,在0、2、4、8、24小时进样至高效液相色谱仪中测定,根据所得色谱图,记录峰面积,计算丹酚酸b的含量,计算rsd,结果如表12所示。
[0416]
表12
[0417][0418]
结果显示,本方法24小时内稳定。
[0419]
3.2.5.6重复性试验
[0420]
取“1”项下的中药组合物适量,按“3.2.2”项下的供试品溶液的制备方法制备6份供试品溶液,按“3.2.4.5”项下的色谱条件,进样至高效液相色谱仪中测定,根据所得色谱图,记录峰面积,计算丹酚酸b的含量,计算rsd,结果如表13所示。
[0421]
表13
[0422][0423]
结果显示,本方法重复性良好。
[0424]
3.2.5.7加样回收试验
[0425]
取“1”项下的中药组合物0.5g,平行6份,精密称定,分别精密加入对照品溶液2、2、4、4、8、8ml,加体积分数为75%的甲醇溶液至刻度,超声30min,取出放置冷却,加体积分数为75%的甲醇溶液补足损失的体积,取1ml用0.45μm的微孔滤头过滤,制备6份供试品溶液,按“3.2.4.5”项下的色谱条件,进样至高效液相色谱仪中测定,根据所得色谱图,记录峰面积,计算丹酚酸b的含量、回收率和rsd值。结果见表14。
[0426]
表14
[0427][0428]
3.2.6三批样品含量测定
[0429]
取3批(批号见表15)待测中药组合物样品0.5g,精密称定,平行2份,按“3.2.2”项下的供试品溶液的制备方法制备待测中药组合物样品溶液,按“3.2.4.5”项下的色谱条件进样至高效液相色谱仪中测定,根据所得色谱图计算丹酚酸b的含量,结果如表15所示。
[0430]
表15
[0431][0432]
结果显示,三批样品中丹酚酸b含量均大于0.25mg/g。
[0433]
3.3黄芪甲苷含量测定
[0434]
3.3.1仪器、试剂
[0435]
仪器:agilent1260高效液相色谱仪;蒸发光检测器(allteach,2000es);色谱柱:thermo ods-2hypersil(150mm
×
4.6mm,5μm);研钵、量筒、圆底烧瓶、冷凝管、蒸发皿等均购于广州市东征化玻仪器有限公司;电热套(巩义市予华仪器有限责任公司);电热套(巩义市予华仪器有限责任公司)、bs224s分析天平(北京赛多利斯仪器系统有限公司)。
[0436]
试药:乙腈(色谱纯);超纯水;正丁醇;甲醇;氨水;黄芪甲苷对照品,待测中药组合物样品(批号:20140801,20140802,20140803,20051211,均由康臣药业(内蒙古)责任有限公司提供)。
[0437]
3.3.2对照品溶液、供试品溶液、阴性样品溶液的制备
[0438]
对照品溶液:精密称取黄芪甲苷对照品适量,加甲醇制成每1ml含黄芪甲苷0.15mg的溶液,即得对照品溶液。
[0439]
供试品溶液:取“1”项下的中药组合物适量研细,取约10g,精密称定,置索氏提取器中,加甲醇100ml,加热回流提取4小时,提取液回收溶剂并浓缩至干,残渣加水20ml,微热使其溶解,用水饱和的正丁醇振摇提取4次,每次40ml,合并正丁醇相,用氨试液充分洗涤2次,每次40ml,弃去氨液,正丁醇相蒸干,残渣用甲醇溶解并转移至5ml量瓶中,加甲醇至刻度,摇匀,即得供试品溶液。
[0440]
阴性样品溶液:取不含黄芪的处方量药材,按“1”项下的中药组合物的制备工艺及“3.3.2”项下的供试品溶液制备方法制备阴性样品溶液。
[0441]
3.3.3色谱条件
[0442]
色谱柱:thermo ods-2hypersil(150mm
×
4.6mm,5μm)
[0443]
流动相:乙腈-水溶液=30:70(v/v),等度洗脱
[0444]
检测器参数:n2流速为2.8ml/min,温度为105℃
[0445]
流速:1ml/min
[0446]
柱温:30℃
[0447]
进样量:10μl
[0448]
3.3.4色谱条件的考察
[0449]
3.3.4.1色谱柱的考察
[0450]
参考《中国药典》2020版一部黄芪甲苷含量测定,其色谱柱选用十八烷基硅烷键合硅胶为填充剂。取“1”项下的中药组合物适量,按“3.3.2”项下的供试品溶液制备方法制备供试品溶液,将所得供试品溶液进样至高效液相色谱仪中测定,其中,除了色谱柱分别为thermo ods-2hypersil、agilent、alltima(规格见表16)外,其他条件按“3.3.3”项下的色谱条件进行,根据所得色谱图观察分离度和理论塔板数,结果如表16所示。
[0451]
表16
[0452][0453]
结果显示:三种色谱柱均达到很好的分离效果,本方法选择thermo ods-2hypersil色谱柱。
[0454]
3.3.4.2流动相的考察
[0455]
以《中国药典》2020版一部黄芪甲苷含量测定方法为基础,取“1”项下的中药组合物适量,按“3.3.2”项下的供试品溶液制备方法制备供试品溶液,将所得供试品溶液进样至高效液相色谱仪中测定,其中,除了流动相分别为体积比30:70的乙腈-水、体积比31:69的乙腈-水、体积比29:71的乙腈-水外,其他条件按“3.3.3”项下的色谱条件进行,根据所得色谱图观察分离度。
[0456]
结果显示:三者差异不显著。本方法选择流动相为体积比30:70的乙腈-水。
[0457]
3.3.4.3柱温的考察
[0458]
取“1”项下的中药组合物适量,按“3.3.2”项下的供试品溶液制备方法制备供试品溶液,将所得供试品溶液进样至高效液相色谱仪中测定,其中,除了柱温分别为25℃、30℃、40℃外,其他条件按“3.3.3”项下的色谱条件进行,根据所得色谱图观察分离度。
[0459]
结果显示:当柱温为30℃时效果最好,选择柱温为30℃。
[0460]
3.3.4.4色谱条件的确认
[0461]
色谱柱:thermo ods-2hypersil(150mm
×
4.6mm,5μm)
[0462]
流动相:乙腈-水溶液=30:70(v/v),等度洗脱
[0463]
检测器参数:n2流速为2.8ml/min,温度为105℃
[0464]
流速:1ml/min
[0465]
柱温:30℃
[0466]
进样量:10μl
[0467]
3.3.5系统适应性研究和方法学验证
[0468]
3.3.5.1系统适应性研究
[0469]
色谱分离:在“3.3.4.4”项下,黄芪甲苷的色谱峰的保留时间约为11min,与其它色谱峰分离良好,分离度大于1.5,对称性为0.97,符合标准。
[0470]
理论塔板数:按公式n=5.54(tr/w
h/2
)计算黄芪甲苷峰的理论塔板数为14294,考虑到不同色谱柱条件(柱长、载体性能、填充情况、流动相比例、使用时间等)差异,暂定黄芪甲苷峰的理论塔板数不小于4000。
[0471]
3.3.5.2专属性试验
[0472]
分别取甲醇空白溶剂、“3.3.2”项下的对照品溶液,“3.3.2”项下的供试品溶液以及“3.3.2”项下的阴性样品溶液各10μl,按“3.3.4.4”项下的色谱条件进样至高效液相色谱仪中测定。
[0473]
结果表明:阴性无干扰。
[0474]
3.3.5.3线性范围考察
[0475]
取“3.3.2”项下的对照品溶液,分别进样4、6、10、20、25μl,按“3.3.4.4”项下的色谱条件进样至高效液相色谱仪中测定,根据所得色谱图,记录峰面积,以浓度为横坐标,峰面积为纵坐标,绘制标准曲线,结果如表17所示,回归方程为:y=0.6667x-4.259(r2=0.9986)。
[0476]
表17
[0477][0478]
结果显示,在0.63~3.9μg范围内线性关系良好。
[0479]
3.3.5.4精密度试验
[0480]
取“3.3.2”项下的对照品溶液,按“3.3.4.4”项下的色谱条件,连续进样至高效液相色谱仪中测定5次,根据所得色谱图,记录峰面积,计算黄芪甲苷的含量,计算rsd,结果如表18所示。
[0481]
表18
[0482][0483]
结果显示,本方法精密度良好。
[0484]
3.3.5.5稳定性试验
[0485]
取“1”项下的中药组合物适量,按“3.3.2”项下的供试品溶液的制备方法制备供试品溶液,按“3.3.4.4”项下的色谱条件,在0、12、15、18、30小时进样至高效液相色谱仪中测定,根据所得色谱图,记录峰面积,计算黄芪甲苷的含量,计算rsd,结果如表19所示。
[0486]
表19
[0487][0488]
结果显示,本方法30小时内稳定。
[0489]
3.3.5.6重复性试验
[0490]
取“1”项下的中药组合物适量,按“3.3.2”项下的供试品溶液的制备方法制备6份供试品溶液,按“3.3.4.4”项下的色谱条件,进样至高效液相色谱仪中测定,根据所得色谱图,记录峰面积,计算黄芪甲苷的含量,计算rsd,结果如表20所示。
[0491]
表20
[0492]
[0493][0494]
结果显示,本方法重复性良好。
[0495]
3.3.5.7加样回收试验
[0496]
取中药组合物(批号20051211),6份每份约0.5g,精密称定,分别份加入黄芪甲苷对照品溶液(0.1569mg/ml)5ml,按制成供试品溶液,按按“3.3.2”项下的供试品溶液的制备方法制备6份供试品溶液,按“3.3.4.4”项下的色谱条件,进样至高效液相色谱仪中测定,根据所得色谱图,记录峰面积,计算黄芪甲苷的含量、回收率和rsd值。结果如表21所示。
[0497]
表21
[0498][0499]
结果显示:回收率在94-103%之间,rsd≤3%。
[0500]
3.3.6三批样品含量测定
[0501]
取3批(批号见表22)待测中药组合物样品各10g,精密称定,平行2份,按“3.3.2”项下的供试品溶液的制备方法制备待测中药组合物样品溶液,按“3.3.4.4”项下的色谱条件进样至高效液相色谱仪中测定,根据所得色谱图计算黄芪甲苷的含量,结果如表22所示。
[0502]
表22
[0503][0504]
结果显示,三批样品中黄芪甲苷含量均大于0.1mg/g。
[0505]
3.4 2,3,5,4
’‑
四羟基二苯乙烯-2-o-β-d葡萄糖苷含量测定
[0506]
3.4.1仪器、试剂
[0507]
仪器:waters2695/2487高效液相色谱仪,tu-1901型紫外分光光度计;色谱柱:phenomenex c
18
(250
×
4.6mm,5μm);kq3200db型数控超声仪(巩义市予华仪器有限责任公司);电热套(巩义市予华仪器有限责任公司)、bs224s分析天平(北京赛多利斯仪器系统有限公司);研钵、量筒、圆底烧瓶、冷凝管、蒸发皿等均购于广州市东征化玻仪器有限公司;电热套(巩义市予华仪器有限责任公司)。
[0508]
试药:乙腈(色谱纯);超纯水;甲酸(色谱纯),乙醇、甲醇、2,3,5,4
’‑
四羟基二苯乙烯-2-o-β-d葡萄糖苷对照品、样品(批号:20140801,20140802,20140803,均由康臣药业(内蒙古)责任有限公司提供)。
[0509]
3.4.2对照品溶液、供试品溶液、阴性样品溶液的制备
[0510]
对照品溶液:称取2,3,5,4
’‑
四羟基二苯乙烯-2-o-β-d葡萄糖苷对照品适量,加稀
乙醇(体积分数为30%的乙醇溶液)定容至20ml量瓶中,超声使完全溶解,制得2,3,5,4
’‑
四羟基二苯乙烯-2-o-β-d葡萄糖苷对照品储备液(浓度为0.6000mg/ml),稀释,制备对照品溶液。
[0511]
供试品溶液:取“1”项下的中药组合物6g,精密称定,置具塞锥形瓶中,精密加入稀乙醇,密塞,称重,超声30min处理,放冷,再称重,用稀乙醇补足减少的重量,摇匀,过滤,即得供试品溶液。
[0512]
阴性样品溶液:取不含制何首乌的处方量药材,按“1”项下的中药组合物的制备工艺及“3.4.2”项下的供试品溶液制备方法制备阴性样品溶液。
[0513]
3.4.3色谱条件
[0514]
色谱柱:dikma c
18
(200
×
4.6mm,5μm)
[0515]
流动相:乙腈-水=15:85(v/v),等度洗脱
[0516]
检测波长:320nm
[0517]
流速:1.0ml/min
[0518]
柱温:25℃
[0519]
进样量:10μl
[0520]
3.4.4供试品溶液制备方法的考察
[0521]
3.4.4.1提取溶剂的考察
[0522]
取“1”项下的中药制剂6g,精密称定,除了提取溶剂分别为稀乙醇、体积分数为70%的乙醇溶液、甲醇外,其他条件按“3.4.2”项下的供试品溶液制备方法进行,制备供试品溶液。将所得供试品溶液和“3.4.2”项下的对照品溶液分别按“3.4.3”项下的色谱条件进样至高效液相色谱仪中测定,根据所得色谱图计算2,3,5,4
’‑
四羟基二苯乙烯-2-o-β-d葡萄糖苷的含量,结果如表23所示。
[0523]
表23
[0524][0525]
结果显示:稀乙醇、体积分数为70%的乙醇溶液差异不大,考虑成本,选择提取溶剂为稀乙醇。
[0526]
3.4.4.2提取时间的考察
[0527]
取“1”项下的中药组合物6g,精密称定,除了超声提取的时间分别为15min、30min、60min外,其他条件按“3.4.2”项下的供试品溶液制备方法进行,制备供试品溶液。将所得供试品溶液和“3.4.2”项下的对照品溶液分别按“3.4.3”项下的色谱条件进样至高效液相色谱仪中测定,根据所得色谱图计算2,3,5,4
’‑
四羟基二苯乙烯-2-o-β-d葡萄糖苷的含量,结果如表24所示。
[0528]
表24
[0529][0530]
结果显示:超声提取30min的提取效率最大,故选择超声提取的时间为30min。
[0531]
3.4.4.3供试品溶液的制备方法的确认
[0532]
取本品6g,精密称定,置具塞锥形瓶中,精密加入稀乙醇,密塞,称重,超声30min处理,放冷,再称重,用稀乙醇补足减少的重量,摇匀,过滤,即得供试品溶液。
[0533]
3.4.5色谱条件的考察
[0534]
3.4.5.1波长的确定
[0535]
取2,3,5,4
’‑
四羟基二苯乙烯-2-o-β-d葡萄糖苷对照品约10mg,加稀乙醇定容至20ml,精密吸取2ml,加稀乙醇定容至10ml,在210~450nm间扫描测定。
[0536]
结果显示:2,3,5,4
’‑
四羟基二苯乙烯-2-o-β-d葡萄糖苷在320nm处有最大吸收。
[0537]
3.4.5.2色谱柱的考察
[0538]
取“1”项下的中药组合物6g,精密称定,按“3.4.4.3”项下的供试品溶液制备方法制备供试品溶液,将所得供试品溶液进样至高效液相色谱仪中测定,其中,除了色谱柱分别为dikma、agilent、kromsil(规格见表25)外,其他条件按“3.4.3”项下的色谱条件进行,根据所得色谱图观察分离度和理论塔板数,结果如表25所示。
[0539]
表25
[0540][0541]
结果显示:dikma色谱柱分离效果最好。
[0542]
3.4.5.3流动相的考察
[0543]
取“1”项下的中药组合物6g,精密称定,按“3.4.4.3”项下的供试品溶液制备方法制备供试品溶液,将所得供试品溶液进样至高效液相色谱仪中测定,其中,除了流动相分别为体积比为15:85的乙腈-水、体积比为14:86的乙腈-水、体积比为16:84的乙腈-水外,其他条件按“3.4.3”项下的色谱条件进行,根据所得色谱图观察分离度。
[0544]
结果显示:最终确定流动相体积比为15:85的乙腈-水。
[0545]
3.4.5.4柱温的考察
[0546]
研究中发现,不同的柱温对待测成分与杂质峰的分离度有一定的影响。取“1”项下的中药组合物6g,精密称定,按“3.4.4.3”项下的供试品溶液制备方法制备供试品溶液,将所得供试品溶液进样至高效液相色谱仪中测定,其中,除了柱温分别为25℃、30℃、40℃外,其他条件按“3.4.3”项下的色谱条件进行,根据所得色谱图观察分离度。
[0547]
结果显示:在一般情况下,随柱温升高,2,3,5,4
’‑
四羟基二苯乙烯-2-o-β-d葡萄糖苷与杂质峰分离度下降。实际检验中应根据样品分离情况在25℃~40℃范围内选择适宜的柱温。本方法选择柱温为25℃。
[0548]
3.4.5.5色谱条件的确认
[0549]
色谱柱:dikma c
18
(200
×
4.6mm,5μm)
[0550]
流动相:乙腈-水=15:85(v/v),等度洗脱
[0551]
检测波长:320nm
[0552]
流速:1.0ml/min
[0553]
柱温:25℃
[0554]
进样量:10μl
[0555]
3.4.6系统适应性研究和方法学验证
[0556]
3.4.6.1系统适应性研究
[0557]
色谱分离:在“3.4.5.5”项下,2,3,5,4
’‑
四羟基二苯乙烯-2-o-β-d葡萄糖苷的色谱峰保留时间约为12min,与其它峰分离良好,分离度大于1.5,对称性为0.96,符合标准。
[0558]
理论塔板数:按公式n=5.54(tr/w
h/2
)计算2,3,5,4
’‑
四羟基二苯乙烯-2-o-β-d葡萄糖苷的理论塔板数为8395,考虑到不同色谱柱条件:柱长、载体性能、填充情况、流动相、使用时间等差异,暂定2,3,5,4
’‑
四羟基二苯乙烯-2-o-β-d葡萄糖苷色谱峰的理论塔板数不小于4000。
[0559]
3.4.6.2专属性试验
[0560]
分别取提取溶剂(空白水溶剂)、“3.4.2”项下的对照品溶液,“3.4.4.3”项下的供试品溶液以及“3.4.2”项下的阴性样品溶液各10μl,按“3.4.5.5”项下的色谱条件进样至高效液相色谱仪中测定。
[0561]
结果表明:阴性无干扰。
[0562]
3.4.6.3线性范围考察
[0563]
取“3.4.2”项下的对照品储备液(浓度为0.6000mg/ml),精密量取对照品储备液0.5、1、2、3、4、5ml,分别置20ml量瓶中,加稀乙醇稀释至刻度,摇匀,分别精密吸取10μl,按“3.4.5.5”项下的色谱条件进样至高效液相色谱仪中测定,根据所得色谱图,记录峰面积,以浓度为横坐标,峰面积为纵坐标,绘制标准曲线,结果如表26所示,回归方程为:y=3528919.5251x+16945.7014
[0564]
表26
[0565][0566]
结果显示,在0.1500~1.5000μg范围内线性关系良好。
[0567]
3.4.6.4精密度试验
[0568]
取浓度为90.000μg/ml的2,3,5,4
’‑
四羟基二苯乙烯-2-o-β-d葡萄糖苷对照品溶液,按“3.4.5.5”项下的色谱条件,连续进样至高效液相色谱仪中测定5次,根据所得色谱图,记录峰面积,计算2,3,5,4
’‑
四羟基二苯乙烯-2-o-β-d葡萄糖苷的含量,计算rsd,结果如表27所示。
[0569]
表27
[0570][0571]
结果表明,本方法精密度良好。
[0572]
3.4.6.5稳定性试验
[0573]
取“1”项下的中药组合物6g,精密称定,按“3.4.4.3”项下的供试品溶液的制备方法制备供试品溶液,按“3.4.5.5”项下的色谱条件,在0、1、2、4、8、16、24小时进样至高效液相色谱仪中测定,根据所得色谱图,记录峰面积,计算2,3,5,4
’‑
四羟基二苯乙烯-2-o-β-d葡萄糖苷的含量,计算rsd,结果如表28所示。
[0574]
表28
[0575][0576][0577]
结果显示,本方法24小时内稳定。
[0578]
3.4.6.6重复性试验
[0579]
取“1”项下的中药组合物6g,精密称定,按“3.4.4.3”项下的供试品溶液的制备方法制备6份供试品溶液,按“3.4.5.5”项下的色谱条件进样至高效液相色谱仪中测定,根据所得色谱图,记录峰面积,计算2,3,5,4
’‑
四羟基二苯乙烯-2-o-β-d葡萄糖苷的含量,计算rsd,结果如表29所示。
[0580]
表29
[0581][0582]
结果显示,本方法重复性良好。
[0583]
3.4.6.7加样回收试验
[0584]
取已知2,3,5,4
’‑
四羟基二苯乙烯-2-o-β-d葡萄糖苷含量的中药组合物3g,分别加入2,3,5,4
’‑
四羟基二苯乙烯-2-o-β-d葡萄糖苷对照品溶液(浓度为0.6360mg/ml)各2ml、2.5ml、3ml,挥干溶剂,按“3.4.4.3”项下的供试品溶液的制备方法制备6份供试品溶液,按“3.4.5.5”项下的色谱条件进样至高效液相色谱仪中测定,根据所得色谱图计算2,3,5,4
’‑
四羟基二苯乙烯-2-o-β-d葡萄糖苷的含量、回收率及rsd值,结果如表30所示。
[0585]
表30
[0586][0587]
3.4.7三批样品含量测定
[0588]
取3批(批号见表31)待测中药组合物样品,6g,精密称定,平行2份,按“3.4.4.3”项下的供试品溶液的制备方法制备待测中药组合物样品溶液,将所得待测中药组合物样品溶液和“3.4.2”项下的对照品溶液分别按“3.4.5.5”项下的色谱条件进样至高效液相色谱仪中测定,根据所得色谱图计算2,3,5,4
’‑
四羟基二苯乙烯-2-o-β-d葡萄糖苷的含量,结果如表31所示。
[0589]
表31
[0590][0591]
结果显示,三批样品中2,3,5,4
’‑
四羟基二苯乙烯-2-o-β-d葡萄糖苷的含量均大于0.1mg/g。
[0592]
3.5苦参碱含量测定
[0593]
3.5.1仪器、试剂
[0594]
仪器:agilent1260高效液相色谱仪,dad检测器;色谱柱:zorbax eclipsec
18
(250
×
4.6mm,5μm);kq3200db型数控超声仪(巩义市予华仪器有限责任公司);电热套(巩义市予华仪器有限责任公司)、bs224s分析天平(北京赛多利斯仪器系统有限公司);研钵、量筒、圆底烧瓶、冷凝管、蒸发皿等均购于广州市东征化玻仪器有限公司;电热套(巩义市予华仪器有限责任公司)。
[0595]
试药:乙腈(色谱纯);超纯水;甲酸(色谱纯),乙醇、甲醇、苦参碱对照品、待测中药组合物样品(批号:20140801,20140802,20140803,均由康臣药业(内蒙古)责任有限公司提供)。
[0596]
3.5.2对照品溶液、供试品溶液、阴性样品溶液的制备
[0597]
对照品溶液:精密称取经五氧化二磷干燥的苦参碱对照品适量,加“3.5.3”项下流动相溶解并制成每1ml约含0.1mg的溶液,即得对照品溶液。
[0598]
供试品溶液:取“1”项下的中药组合物适量,研细,取约2g,精密称定,置具塞锥形瓶中,加水25ml和氨水0.1ml,超声使完全溶解,溶液转移至分液漏斗中,用少量水洗涤容器,洗涤液一并转移到分液漏斗中,加三氯甲烷溶液萃取3次,每次15ml,合并三氯甲烷萃取
液,减压回收溶剂,残渣用“3.5.3”项下流动相溶解,转移至10ml量瓶中,并用“3.5.3”项下流动相稀释至刻度,摇匀,滤过,取续滤液,即得供试品溶液。
[0599]
阴性样品溶液:取不含苦参的处方量药材,按“1”项下的中药组合物的制备工艺及“3.5.2”项下的供试品溶液制备方法制备阴性样品溶液。
[0600]
3.5.3色谱条件
[0601]
色谱柱:agilent zorbax eclipse c
18
(250
×
4.6mm,5μm)
[0602]
流动相:乙腈-体积分数为0.1%的磷酸溶液(用三乙胺调节流动相的ph值至8.0
±
0.1)=20:80(v/v),等度洗脱
[0603]
检测波长:220nm
[0604]
流速:1.0ml/min
[0605]
柱温:25℃
[0606]
进样量:10μl
[0607]
3.5.4供试品溶液制备方法的考察
[0608]
3.5.4.1氨水用量的考察
[0609]
取“1”项下的中药组合物适量,研细,取约2g,精密称定,除了氨水的加入量分别为0.1ml、0.3ml、0.5ml外,其他条件按“3.5.2”项下的供试品溶液制备方法进行,制备供试品溶液。将所得供试品溶液和“3.5.2”项下的对照品溶液分别按“3.5.3”项下的色谱条件进样至高效液相色谱仪中测定,根据所得色谱图计算苦参碱的含量,结果如表32所示。
[0610]
表32
[0611][0612]
结果显示,氨水加入量为0.1ml时,苦参碱含量最高。
[0613]
3.5.4.2水用量的考察
[0614]
取“1”项下的中药组合物适量,研细,取约2g,精密称定,除了水的加入量分别为25ml、50ml、100ml外,其他条件按“3.5.2”项下的供试品溶液制备方法进行,制备供试品溶液。将所得供试品溶液和“3.5.2”项下的对照品溶液分别按“3.5.3”项下的色谱条件进样至高效液相色谱仪中测定,根据所得色谱图计算苦参碱的含量,结果如表33所示。
[0615]
表33
[0616][0617]
结果显示,水加入量为25ml时,苦参碱含量最高。
[0618]
3.5.4.3三氯甲烷萃取次数的考察
[0619]
取“1”项下的中药组合物适量2份,研细,取约2g,精密称定,除了三氯甲烷溶液萃取3次后,每次均收集三氯甲烷相外,其他条件按“3.5.2”项下的供试品溶液制备方法进行,制备供试品溶液。将所得供试品溶液和“3.5.2”项下的对照品溶液分别按“3.5.3”项下的色谱条件进样至高效液相色谱仪中测定,根据所得色谱图计算苦参碱的含量,结果如表34所
示。
[0620]
表34
[0621][0622]
结果显示,三次萃取总和时,苦参碱含量最高。
[0623]
3.5.4.4三氯甲烷萃取用量的考察
[0624]
取“1”项下的中药组合物适量4份,研细,取约2g,精密称定,除了三氯甲烷溶液三次萃取时的用量分别为10ml、10ml、10ml,15ml、15ml、15ml,20ml、20ml、20ml,25ml、25ml、25ml外,其他条件按“3.5.2”项下的供试品溶液制备方法进行,制备供试品溶液。将所得供试品溶液和“3.5.2”项下的对照品溶液分别按“3.5.3”项下的色谱条件进样至高效液相色谱仪中测定,根据所得色谱图计算苦参碱的含量,结果如表35所示。
[0625]
表35
[0626][0627]
结果显示,每次用量为15、20、25ml时,含量无明显差异,为控制检验成本,因此选择萃取用量为15ml。
[0628]
3.5.4.5供试品溶液的制备方法的确认
[0629]
取本品2g,精密称定,置具塞锥形瓶中,加水25ml和氨水0.1ml,超声使完全溶解,溶液转移至分液漏斗中,用少量水洗涤容器,洗涤液一并转移到分液漏斗中,加三氯甲烷溶液萃取3次,每次15ml,合并三氯甲烷萃取液,减压回收溶剂,残渣用“3.5.3”项下流动相溶解,转移至10ml量瓶中,并用“3.5.3”项下流动相稀释至刻度,摇匀,滤过,取续滤液,即得供试品溶液。
[0630]
3.5.5色谱条件的考察
[0631]
3.5.5.1波长的确定
[0632]
参考2020年版《中国药典》,本方法选择检测波长为220nm。
[0633]
3.5.5.2色谱柱的考察
[0634]
取“1”项下的中药组合物适量,研细,取约2g,精密称定,按“3.5.4.5”项下的供试品溶液制备方法制备供试品溶液,将所得供试品溶液进样至高效液相色谱仪中测定,其中,除了色谱柱分别为agilent、ultimate、agilent(规格见表36)外,其他条件按“3.5.3”项下的色谱条件进行,根据所得色谱图观察分离度和理论塔板数,结果如表36所示。
[0635]
表36
[0636][0637]
结果显示:注意到苦参碱色谱峰前峰的情况,ultimate xb-c18因苦参碱色谱峰包含了杂质峰,未到达分离要求,测得结果偏高,agilent zorbax eclipse色谱柱可以满足苦参碱含量测定要求。
[0638]
3.5.5.3流动相的考察
[0639]
取“1”项下的中药组合物适量,研细,取约2g,精密称定,按“3.5.4.5”项下的供试品溶液制备方法制备供试品溶液,将所得供试品溶液进样至高效液相色谱仪中测定,其中,除了流动相ph值分别为7.8、8.0、8.2外,其他条件按“3.5.3”项下的色谱条件进行,根据所得色谱图记录峰面积,计算苦参碱含量,结果如表37所示。
[0640]
表37
[0641][0642]
结果显示,流动相ph值的微小改变对苦参碱含量测定没有影响。当ph值为7.8时,色谱峰的对称性明显降低,ph值升高,有利于色谱峰的分离,但对色谱柱损伤较大,故确定流动相ph值为8.0
±
0.1。
[0643]
3.5.5.4柱温的考察
[0644]
取“1”项下的中药组合物适量,研细,取约2g,精密称定,按“3.5.4.5”项下的供试品溶液制备方法制备供试品溶液,将所得供试品溶液进样至高效液相色谱仪中测定,其中,除柱温分别为20℃、25℃、30℃外,其他条件按“3.5.3”项下的色谱条件进行,根据所得色谱图记录峰面积,计算苦参碱含量,结果如表38所示。
[0645]
表38
[0646][0647]
结果显示,柱温的微小改变对苦参碱含量测定没有影响。本方法选择柱温为25℃。
[0648]
3.5.5.5流速的考察
[0649]
取“1”项下的中药组合物适量,研细,取约2g,精密称定,按“3.5.4.5”项下的供试品溶液制备方法制备供试品溶液,将所得供试品溶液进样至高效液相色谱仪中测定,其中,除流速分别为0.8ml/min、1.0ml/min、1.2ml/min外,其他条件按“3.5.3”项下的色谱条件进行,根据所得色谱图记录峰面积,计算苦参碱含量,结果如表39所示。
[0650]
表39
[0651][0652]
结果显示,流速的微小改变对苦参碱含量测定没有影响。本方法选择流速1.0ml/min。
[0653]
3.5.5.6色谱条件的确认
[0654]
色谱柱:agilent zorbax eclipse c
18
(250
×
4.6mm,5μm)
[0655]
流动相:乙腈-体积分数为0.1%的磷酸溶液(用三乙胺调节流动相的ph值至8.0
±
0.1)=20:80(v/v),等度洗脱
[0656]
检测波长:220nm
[0657]
流速:1.0ml/min
[0658]
柱温:25℃
[0659]
进样量:10μl
[0660]
3.5.6系统适应性研究和方法学验证
[0661]
3.5.6.1系统适应性研究
[0662]
色谱分离:在“3.5.5.6”项下,苦参碱的色谱峰保留时间约为15min,与其它峰分离良好,分离度大于1.5,对称性为1.06,符合标准。
[0663]
理论塔板数:按公式n=5.54(tr/w
h/2
)计算苦参碱的理论塔板数为11849,考虑到不同色谱柱条件:柱长、载体性能、填充情况、流动相、使用时间等差异,暂定苦参碱色谱峰的理论塔板数不小于4000。
[0664]
3.5.6.2专属性试验
[0665]
分别取提取溶剂(氨水和水混合溶剂)、“3.5.2”项下的对照品溶液,“3.5.4.5”项下的供试品溶液以及“3.5.2”项下的阴性样品溶液各10μl,按“3.5.5.6”项下的色谱条件进样至高效液相色谱仪中测定。
[0666]
结果显示:阴性无干扰。
[0667]
3.5.6.3线性范围考察
[0668]
精密吸取苦参碱对照品溶液(浓度为0.1096mg/ml)2、4、8、12、16、20μl,按“3.5.5.6”项下的色谱条件进样至高效液相色谱仪中测定,根据所得色谱图,记录峰面积,以浓度为横坐标,峰面积为纵坐标,绘制标准曲线,结果如表40所示,回归方程为:y=807.42x-2.2416,r2=1。
[0669]
表40
[0670][0671]
结果显示,0.2192~2.192μg范围内线性关系良好。
[0672]
3.5.6.4中间精密度试验
[0673]
取“1”项下的中药组合物适量,研细,取约2g,精密称定,按“3.5.4.5”项下的供试品溶液的制备方法制备供试品溶液6份,甲、乙、丙不同人员在不同日期、不同仪器上,分别按“3.5.5.6”项下的色谱条件进样至高效液相色谱仪中测定,根据所得色谱图,记录峰面积,计算苦参碱的含量,计算rsd,结果如表41所示。
[0674]
表41
[0675][0676]
结果显示,本方法中间精密度良好。
[0677]
3.5.6.5稳定性试验
[0678]
取“1”项下的中药组合物适量,研细,取约2g,精密称定,按“3.5.4.5”项下的供试品溶液的制备方法制备供试品溶液,按“3.5.5.6”项下的色谱条件,在0、2、4、6、8小时进样至高效液相色谱仪中测定,根据所得色谱图,记录峰面积,计算苦参碱的含量,计算rsd,结果如表42所示。
[0679]
表42
[0680][0681]
结果显示,本方法8小时内稳定。
[0682]
3.5.6.6重复性试验
[0683]
取“1”项下的中药组合物适量,研细,取约2g,精密称定,按“3.5.4.5”项下的供试品溶液的制备方法制备供试品溶液6份,按“3.5.5.6”项下的色谱条件进样至高效液相色谱仪中测定,根据所得色谱图,记录峰面积,计算苦参碱的含量,计算rsd,结果如表43所示。
[0684]
表43
[0685][0686]
结果显示,本方法重复性良好。
[0687]
3.5.6.7加样回收试验
[0688]
取“1”项下的中药组合物适量(批号20140616,含量按0.663mg/g计算,取样6份),研细,取约1g,并精密加入对照品溶液(浓度0.769mg/ml)0.9ml,按“3.5.4.5”项下的供试品溶液的制备方法制备供试品溶液6份,按“3.5.5.6”项下的色谱条件进样至高效液相色谱仪
中测定,根据所得色谱图,记录峰面积,计算苦参碱的含量、回收率及rsd值,结果如表44所示。
[0689]
表44
[0690][0691]
结果显示,6份样品的回收率在97.36~100.69%之间,rsd%为1.426,表明回收率结果良好。
[0692]
3.5.7三批样品含量测定
[0693]
取3批(批号见表45)待测中药组合物样品各2g,按“3.5.4.5”项下的供试品溶液的制备方法制备待测中药组合物样品溶液,按“3.5.5.6”项下的色谱条件进样至高效液相色谱仪中测定,根据所得色谱图计算苦参碱的含量,结果如表45所示。
[0694]
表45
[0695][0696]
结果显示,三批样品中苦参碱的含量均大于0.3mg/g。
[0697]
4性状检测
[0698]
4.1性状:采用目测、鼻嗅及口尝,质量指标为:本品为棕色或棕褐色颗粒;味甘、微苦。
[0699]
4.2参照《中国药典》2020年版四部通则颗粒剂项下要求检查,质量指标为应符合《中国药典》2020年版四部通则颗粒剂项下有关的各项规定。
[0700]
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
[0701]
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1