一种白花泡桐叶的HPLC指纹图谱鉴别方法
一种白花泡桐叶的hplc指纹图谱鉴别方法
技术领域
1.本发明属于中药材质量控制技术领域,具体涉及一种白花泡桐叶的hplc指纹图谱鉴别方法。
背景技术:
2.白花泡桐(paulownia fortunei (seem.) hemsl.)为玄参科泡桐属落叶乔木,在我国10多个省市均有分布。白花泡桐作为一种优质的速生木材,其除了广泛地应用于工农业生产外,还是一种常用的中药材,泡桐的花、叶、皮、根、果均可入药。本草纲目对白花泡桐各个部位的药理作用做了详细记载,现代医学研究表明白花泡桐具有抑菌,消炎,抗肿瘤甚至还有杀虫的作用。白花泡桐树枝繁叶茂,叶片的数量巨大,是一笔丰富的资源。用白花泡桐叶作饲料,不仅能促进动物生长,而且能提高动物抗病能力。医疗方面的价值可以体现在治疗痈疽、疔疮、创伤出血等,白花泡桐叶具有重要的经济价值和社会价值。
3.由于泡桐属植物叶片相似度高,特别是中药材经过炮制、切片和打成粉末后通过肉眼难以区分,目前并没有有效的方法对炮制、切片和打成粉末后的白花泡桐叶进行鉴定。
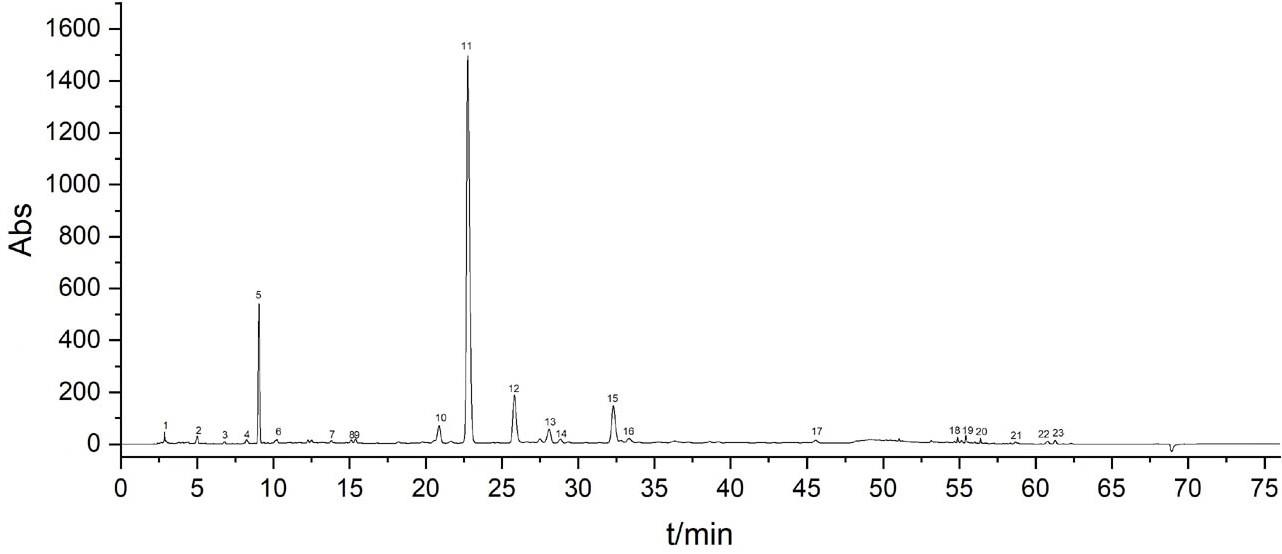
技术实现要素:
4.为克服现有技术的不足,本发明通过大量研究,提出一种白花泡桐叶的hplc指纹图谱鉴别方法。
5.本发明的白花泡桐叶hplc指纹图谱鉴别方法采取以下步骤:(1)样品溶液的制备:分别称取白花泡桐叶粉末和待鉴别样品粉末0.5 g,再分别置具塞锥形瓶中,精密加入70%甲醇10 ml,密塞,称定质量,超声处理(功率500 w,频率40 khz)30 min,放冷,用70%甲醇补足减失的质量,取上清液用 0.45 μm微孔滤膜滤过,分别制得白花泡桐叶样品溶液和待鉴别样品溶液。
6.(2)hplc图谱的收集:分别对白花泡桐叶样品溶液和待鉴别样品溶液进行hplc检测并记录其指纹图谱,色谱条件为:色谱柱:agilent zorbax hc-c18(2) (4.6 mm
×
250 mm,5μm);流动相:甲醇(b)-0.1%甲酸缓冲溶液(a);梯度洗脱:时间变化(min)0-8-44-52-65-66-76、流动相a(%)90-68-50-0-0-90-90、流动相b(%)10-32-50-100-100-10-10;检测波长:254 nm;流速:1 ml/min;柱温:30 ℃;进样量:10 μl。
7.(3)比对图谱:白花泡桐叶样品的hplc特征指纹图谱见图1,图谱中呈现23个特征峰,并应与白花泡桐叶hplc特征指纹图谱的23个特征峰相对应,且峰14与白花泡桐叶样品hplc特征指纹图谱的峰14保留时间一致的待鉴别样品,认定为白花泡桐叶,否则不能被认定为白花泡桐叶。
8.本发明是建立在对白花泡桐叶提取液的hplc色谱条件和指纹谱图大量研究的基础上的,耗费了大量的精力和创造性劳动,其过程如下:色谱柱考察:分别考察了以下的三种色谱柱进行了比较,实验结果表明,1号色谱柱中,各峰分离度较好,色谱峰多且色谱峰形相对较好。1号柱子:agilent zorbax hc-c18
(2) (4.6 mm
×
250 mm,5μm);2号柱子:waters xbridge c18 (4.6 mm
×
150 mm,3.5
µ
m);3号柱子:wondacract c-18wrs (4.6 mm
×
250 mm,5
µ
m)。
9.流动相系统考察:考察了甲醇-水系统、甲醇-0.1%甲酸系统、甲醇-0.1%乙酸、甲醇-0.1%磷酸系统对于白花泡桐叶hplc色谱图的影响。实验研究表明了甲醇-水系统、甲醇-0.1%乙酸、甲醇-0.1%磷酸系统很多色谱峰都无法分离而且重叠色谱峰多。结果表明甲醇-0.1%甲酸系统的分离效果是最好的,色谱峰形好,色谱出峰的时间合适。因此选择甲醇-0.1%甲酸系统。
10.检测波长考察:分别考察在以下的这六个不同波长下的指纹图谱,为210 nm、254 nm、270nm、290 nm、320 nm、365 nm。实验研究表明在254 nm 波长下的各个色谱峰均具有较好的紫外光吸收,其色谱信息较丰富,基线较平稳,故选择254 nm作为白花泡桐叶的测定波长。
11.柱温考察:分别考察了 25 ℃,30 ℃,35℃下的色谱图,在其它的色谱条件一致的情况下,在 30 ℃的条件下,各峰的分离效果最佳,因此,确定柱温为 30 ℃。
12.同时,通过大量研究确定流速为1 ml/min;进样量:10 μl。
13.同时,在大量试验中发现白花泡桐叶hplc指纹图谱中的11号峰为共有峰,强度较高,保留时间相对居中与其余色谱峰的分离度良好,因此选定11号峰为参照峰。因不确定该峰所代表的化合物,对其进行快速制备及鉴定。
14.称取白花泡桐叶粉末适量,置于具塞锥形瓶中,加入70%甲醇提取,超声处理(功率500 w,频率40 khz)30 min,即得提取液。经半制备液相以甲醇-水(33:67)(tr=30.5 min)得到参照峰化合物。
15.参照峰的制备及鉴定:对)参照峰(峰11)进行快速制备,并利用核磁共振光谱仪进行定性,最终核磁定性鉴定为毛蕊花糖苷。制备色谱条件为:色谱柱ymc-pack ph (10 mm
×
250 mm,5
ꢀµ
m);等度洗脱;流动相:甲醇(b)-水(a)=(33:67)。检测波长254 nm;流动相流速2.5 ml/min;柱温30 ℃;tr=24.9 min;岛津lc-20at。
16.制备成分的鉴定:淡黄色粉末。esi-ms m/z: 647.1940 [m+na]
+
。1h nmr (600 mhz, meod) δ: 7.59 (1h, d, j = 16.2 hz, h-7'), 7.05 (1h , d, j = 1.8 hz, h-2'), 6.95 (1h, dd, j = 7.8, 1.8 hz, h-6'), 6.77 (1h, d, j = 7.8 hz, h-5'), 6.69 (1h, d, j = 2.4 hz, h-2), 6.67 (1h, d, j = 7.8 hz, h-5), 6.56 (1h, dd, j = 8.0, 1.8 hz, h-6), 6.27 (1h, d, j = 15.6 hz, h-8'), 5.18 (1h, d, j = 1.8 hz, h-1
″
'), 4.37 (1h, d, j = 7.8 hz, h-1
″
), 4.05 (1h, m, h-8β), 3.71 (1h, m, h-8α), 2.79 (2h, m, h-7), 1.09 (3h, d, j = 6.0 hz, h-6
″
').
13
c-nmr (meod, 125 mhz) δ: 166.9 (c-9'), 148.5 (c-4'), 146.7 (c-7'), 145.5 (c-3'), 144.8 (c-3), 143.3 (c-4), 130.0 (c-1), 126.2 (c-1'), 121.9 (c-6'), 119.8 (c-6),115.7 (c-2), 115.1 (c-5'), 114.9 (c-5), 113.7 (c-8'), 113.2 (c-2'), 102.8 (c-1
″
), 101.7(c-1
″
'), 80.3 (c-3
″
), 74.8 (c-2
″
),74.6 (c-5
″
),72.4 (c-4
″
'), 71.0 (c-2
″
'), 70.9 (c-3
″
'), 70.6 (c-5
″
'), 69.1 (c-4
″
), 69.0 (c-5
″
'), 60.9 (c-6
″
), 35.2 (c-7), 17.0 (c-6
″
')。以上数据与文献报道基本一致[wang xq, li cf, jiang ll, et al. phenylethaniod glycosides from orobanchepycnostachya hance and their chemotaxonomic significance[j]. biochemical systematics andecology,https://
doi.org/10.1016/j.bse.2020.104168.],故鉴定为毛蕊花糖苷。
[0017]
白花泡桐叶hplc特征指纹图谱研究如下:精密度实验:精密称取白花泡桐叶粉末按本发明方法连续进样6次,每次进样量10
ꢀµ
l,记录色谱图。以11号峰(毛蕊花糖苷)为参照峰,考察各共有峰的相对保留时间与相对峰面积。结果表明,各共有峰相对保留时间rsd均小于0.15%,相对峰面积rsd均小于2.44%,表明仪器精密度良好。
[0018]
重复性实验:取同一批白花泡桐叶粉末(s9),按本发明方法平行制备6份供试品溶液,分别进样10
ꢀµ
l,记录色谱图。以11号峰(毛蕊花糖苷)为参照峰,考察各共有峰的相对保留时间与相对峰面积。结果表明,各共有峰相对保留时间rsd均小于0.80%,相对峰面积rsd均小于2.71%,表明方法重复性良好。
[0019]
稳定性实验:精密称取白花泡桐叶粉末0.5 g,按本发明方法分别在 0、2、4、8、12、24 h 进样,每次进样10
ꢀµ
l,记录色谱图。以11号峰(毛蕊花糖苷)为参照峰,考察各共有峰的相对保留时间与相对峰面积。结果表明,各共有峰相对保留时间rsd均小于1.69%,相对峰面积rsd均小于3.51%,表明24 h内供试品溶液的稳定性良好。
[0020]
本发明条件下,对19批白花泡桐叶进行测定,记录各批样品的指纹图谱。采用《中药色谱指纹图谱相似度评价系统(2012 a版)》对19批白花泡桐叶的指纹图谱进行分析,并筛选出重复性好的色谱峰作为共有峰,分别计算各样品指纹图谱中各共有峰与内标峰的相对峰面积,作为各样品指纹图谱数据见图1和图2。供试品特征图谱中应呈现23个特征峰,并应与对照药材参照物色谱峰中的23个特征峰相对应,其中峰11应与参照物峰保留时间一致。
[0021]
白花泡桐叶hplc指纹图谱相似度评价:采用《中药色谱指纹图谱相似度评价系统(2012 a版)》对采集的19批白花泡桐叶进行相似度评价,计算各批次指纹图谱与对照指纹图谱(r)之间的相似度,结果显示,各批次白花泡桐叶指纹图谱与对照指纹图谱(r)的相似度在0.924~1之间。
[0022]
与现有技术相比,本发明的有益效果是:本发明首次利用hplc指纹图谱对白花泡桐叶进行质量控制,该方法能稳定、精密、重现地对白花泡桐叶进行质量控制和有效鉴别。
[0023]
本发明通过对19个不同批次的白花泡桐叶进行检测,结果表明,相同位置的色谱峰的相对保留时间的rsd均在4%以内,说明本发明提供的指纹图谱具有姣好的重现性。本发明提供的白花泡桐叶指纹图谱的建立方法可靠性较高;而且本发明由上述方法建立的白花泡桐叶指纹图谱,具有23个达到有效分离的共有峰,所述指纹图谱能够有效地表征白花泡桐叶的质量,有利于全面监控白花泡桐叶的质量。其与对照图谱的相似度均大于0.99,指纹图谱的分离度和峰形较好。建立了白花泡桐叶指纹图谱检测标准。本发明首次对白花泡桐叶70%甲醇提取物中可以作为参照峰的物质进行制备鉴定,对参照峰进行了指认,填补多项技术空白。
附图说明
[0024]
图1是白花泡桐叶的hplc指纹图谱。
[0025]
图2是19批白花泡桐叶的hplc比对图谱。
[0026]
图3是11号峰(毛蕊花糖苷)的核磁共振h谱。
[0027]
图4是11号峰(毛蕊花糖苷)的核磁共振c谱。
[0028]
图5是白花泡桐、兰考泡桐、毛泡桐和台湾泡桐四种泡桐叶hplc比较图谱。
[0029]
图6是11号峰(毛蕊花糖苷)的制备色谱图。
具体实施方式
[0030]
现结合具体实施例,对本发明进一步具体说明。
[0031]
白花泡桐、兰考泡桐、毛泡桐、台湾泡桐的hplc指纹图谱鉴定:(1)样品溶液的制备:分别称取白花泡桐叶粉末和待鉴别样品粉末0.5 g,再分别置具塞锥形瓶中,精密加入70%甲醇10 ml,密塞,称定质量,超声处理(功率500 w,频率40 khz)30 min,放冷,用70%甲醇补足减失的质量,取上清液用 0.45 μm微孔滤膜滤过,分别制得白花泡桐叶样品溶液和待鉴别样品溶液。
[0032]
(2)hplc图谱的收集:分别对白花泡桐叶样品溶液和待鉴别样品溶液进行hplc检测并记录其指纹图谱,色谱条件为:色谱柱:agilent zorbax hc-c18(2) (4.6 mm
×
250 mm,5μm);流动相:甲醇(b)-0.1%甲酸缓冲溶液(a);梯度洗脱:时间变化(min)0-8-44-52-65-66-76、流动相a(%)90-68-50-0-0-90-90、流动相b(%)10-32-50-100-100-10-10;检测波长:254 nm;流速:1 ml/min;柱温:30 ℃;进样量:10 μl。
[0033]
(3)比对图谱:白花泡桐叶样品的hplc特征指纹图谱见图1,图谱中呈现23个特征峰,并应与白花泡桐叶hplc特征指纹图谱的23个特征峰相对应,且峰11与白花泡桐叶样品hplc特征指纹图谱的峰11保留时间一致的待鉴别样品,认定为白花泡桐叶,否则不能被认定为白花泡桐叶。
[0034]
本发明条件下,白花泡桐、兰考泡桐、毛泡桐、台湾泡桐色谱图见图5。从四种泡桐对比图谱比较来看,在保留时间29.5 min左右,即白花泡桐叶hplc对照图谱中的特征峰有区别于其他三种泡桐的色谱峰,兰考泡桐、毛泡桐、台湾泡桐的hplc指纹图谱均不符合白花泡桐叶hplc特征指纹图谱的认定条件,将白花泡桐叶区分出来。
[0035]
上述只是本发明的较佳实施例,并非对本发明作任何形式上的限制。任何熟悉本领域的技术人员,在不脱离本发明技术方案范围的情况下,都可利用上述揭示的技术内容对本发明技术方案做出许多可能的变动和修饰,或修改为等同变化的等效实施例。因此,凡是未脱离本发明技术方案的内容,依据本发明技术实质对以上实施例所做的任何简单修改、等同变化及修饰,均应落在本发明技术方案保护的范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1