同时测定6种维生素A族化合物的高效液相色谱检测方法
同时测定6种维生素a族化合物的高效液相色谱检测方法
技术领域
1.本发明涉及一种高效液相色谱-二极管阵列检测法同时测定6种维生素a族化合物的方法,属于化学检测技术领域。
背景技术:
2.维生素a是最早被发现的一种脂溶性的维生素,结构组成包括一个β-紫罗酮环、一个由四个头尾相连的类异戊二烯单元组成的侧链以及在碳-15位结合了一个烃基(视黄醇)、或者醛基(视黄醛)、或者羧基(视黄酸)或者酯基(视黄酯)。维生素a一族具有视黄醇生物活性,包括两大类物质,一种是类视黄醇物质,指视黄醇、其代谢产物以及具有相似结构的合成类似物,来源于动物性食物。另一种是维生素a原类胡萝卜素,是膳食视黄醇的前体物质,来自于植物性食物,主要包括β-胡萝卜素、α-胡萝卜素和β-隐黄质,其中β-胡萝卜素的维生素a活性最高,维生素a作为人体必须的一种维生素,对于机体的正常代谢具有重要意义。
3.目前,关于维生素a族化合物的研究多采用皂化的前处理方法,仅能实现对单一视黄醇定量,液相色谱-串联质谱的检测方法可以实现对不同形式维生素a族化合物的定性,但是相较于液相色谱的检测方法检测成本高昂,操作要求严格等特点,目前的检测方法具有一定的局限性。
技术实现要素:
4.本发明提供了一种同时测定6种维生素a族化合物的高效液相色谱检测方法,以弥补现有技术的不足。
5.为达到上述目的,本发明所采取的具体技术方案为:
6.一种同时测定6种维生素a族化合物的高效液相色谱检测方法,包括以下步骤:
7.(1)样品前处理,进行皂化和非皂化处理;
8.(2)配置标准品溶液,包括维生素a、维生素a酸、全反式视黄醛、维生素a醋酸酯、维生素a棕榈酸酯和β-胡萝卜素的标准品;
9.(3)样品溶液和标准品溶液进行高效液相色谱处理;高效液相色谱条件为:色谱柱采用c
30
,梯度洗脱程序为:0-8min 90%a(甲醇)、1%b(超纯水)、9%c(甲基叔丁基醚);8.1-16min35%a(甲醇)、65%c(甲基叔丁基醚);
10.(4)制作维生素a、维生素a酸、全反式视黄醛、维生素a醋酸酯、维生素a棕榈酸酯和β-胡萝卜素的标准品的标准曲线;
11.(5)基于标准曲线,对样品中维生素a族化合物的定量和定性分析。
12.进一步的,所述步骤(1)具体为:
13.1)皂化样品前处理过程为:样品匀浆,加入抗坏血酸和氢氧化钾溶液,将无水乙醇溶液加入匀浆样品中进行超声提取,加入乙酸乙酯萃取,将萃取液经无水硫酸钠干燥后,再减压蒸发,氮干,最后用乙醇、三氯甲烷(1:1,v/v)混合溶液溶解后,有机膜过滤后进高效液
相色谱仪进行分析;
14.2)非皂化样品前处理过程为:样品匀浆,加入抗坏血酸,将无水乙醇溶液加入匀浆样品中进行超声提取,加入石油醚萃取,将萃取液用蒸馏水水洗至萃取液经ph试纸检测呈中性,经无水硫酸钠干燥后,将萃取液进行减压蒸发,氮干,用乙醇、三氯甲烷(1:1,v/v)混合溶液溶解后,有机膜过滤后进高效液相色谱仪进行分析。
15.进一步的,所述步骤(2)中标准品的制备:
16.称取维生素a、维生素a酸、全反式视黄醛、维生素a醋酸酯、维生素a棕榈酸酯标准品分别用抗坏血酸乙醇溶液溶解,得到浓度为1mg/ml的标准储备液,避光低温保存;称取β-胡萝卜素用l抗坏血酸三氯甲烷溶解,得到浓度为1mg/ml的标准储备液。
17.进一步的,所述步骤(3)中,所述高效液相色谱分析条件具体如下:
18.色谱柱采用ymc-βcarotene c
30
(4.6mmx250 mm,3μm),柱温为30-40℃、进样量为20μl、流速为0.8-1.0ml/min、检测波长为325nm、350nm、380nm、460nm,梯度洗脱程序为:梯度洗脱程序为:0-8min 90%a(甲醇)、1%b(超纯水)、9%c(甲基叔丁基醚);8.1min 35%a(甲醇)、65%c(甲基叔丁基醚);8.1-16min 35%a(甲醇)、65%c(甲基叔丁基醚)。
19.进一步的,所述步骤(4)中,分别准确吸取1mg/ml维生素a、维生素a酸、全反式视黄醛、维生素a醋酸酯、维生素a棕榈酸酯、β-胡萝卜素标准工作液200μl,用抗坏血酸乙醇溶液稀释定容配置成浓度分别为50μg/ml、25μg/ml、10μg/ml、5μg/ml、2.5μg/ml、1μg/ml、0.5μg/ml的混合标准工作液,从高浓度到低浓度依次进高效液相色谱仪进行分析测定,以浓度(c)为横坐标,以对应峰面积(a)为纵坐标,进行标准曲线的绘制。
20.与现有技术相比,本发明的优点和有益效果为:
21.(1)本发明采用高效液相色谱-二极管阵列检测法能够实现6种维生素a族化合物同时分离测定,对海洋贝类中的维生素a族化合物进行准确的定量,与高效液相色谱-质谱检测法相比检测费用低,操作简单方便,对仪器要求低。本发明通过对6种维生素a族化合物进行紫外全波长扫描确定了其最大吸收波长为325nm、350nm、380nm、460nm,将其作为检测波长进行分析测定。本发明中选用的c
30
色谱柱适用于类胡萝卜素化合物的分离,对维生素a族化合物也能实现较好的分离效果,选用的甲基叔丁基醚流动相极性小,对酯类物质和β-胡萝卜素的洗脱速度快,所建立梯度洗脱方法能够使得6种维生素a族化合物得到有效的分离,出峰时间稳定且峰型良好,易于进行含量的测定。
22.(2)专属性强
23.本发明建立的方法能够实现对海产品中不同形式的维生素a族化合物的含量测定,且与其他物质间存在相互干扰,实验过程中所用的空白溶剂(抗坏血酸乙醇三氯甲烷混合溶液)对6种维生素a族化合物的检测也无干扰。
24.(3)灵敏度高
25.本发明建立的方法,维生素a、维生素a醋酸酯、维生素a棕榈酸酯、维生素a酸、全反式维生素a醛、β-胡萝卜素的检出限浓度(s/n=3)分别为0.11μg/ml、0.14μg/ml、0.12μg/ml、0.11μg/ml、0.15μg/ml、0.09μg/ml,定量限(s/n=10)分别为0.37μg/ml、0.47μg/ml、0.40μg/ml、0.35μg/ml、0.43μg/ml、0.28μg/ml。
26.(4)实用性强
27.本发明建立的方法能够同时测定6种不同形式的维生素a族化合物,也可适用于不
同海产品中不同形式维生素a族化合物的分析测定。
28.(5)检测速度快
29.本发明尝试了多种不同的流动相,不断优化梯度洗脱条件,提高了分离效率,能够在15分钟内完成6种不同形式维生素a类化合物的分离测定,大大缩短了检测时间,节省了检测时间和检测试剂,经济效益高。本发明提供了一种检测快速、准确度高、分离效果好,且能同时测定6种不同维生素a族化合物的高效液相色谱-二极管阵列检测方法,该方法能够对海洋贝类中不同形式的维生素a族化合物实现准确定性和定量分析。
附图说明
30.图1是6种维生素a族化合物混合溶液对照品分离色谱图。
31.图2是采用乙腈、超纯水、甲基叔丁基醚作为流动相时六种维生素a族化合物分离色谱图。
32.图3是采用乙腈10%甲醇、0.01%磷酸溶液、甲基叔丁基醚作为流动相时维生素a族化合物分离色谱图。
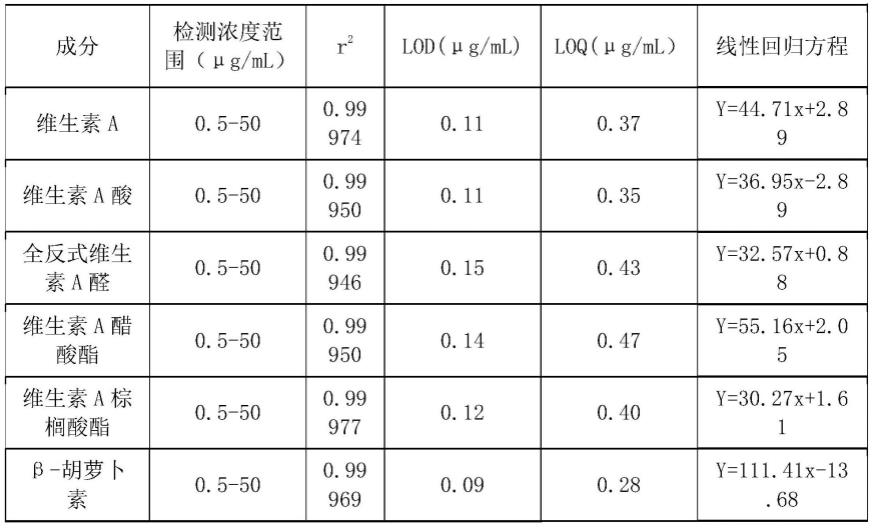
33.图4是采用甲醇、0.01%磷酸溶液、甲基叔丁基醚作为流动相时六种维生素a族化合物分离色谱图。
具体实施方式
34.下面结合具体实例来描述本发明,本发明的优点和特点将会随着描述更加清楚。
35.实施例1:
36.一种同时测定6种维生素a族化合物的高效液相色谱检测方法,包括以下步骤:
37.(1)对照品溶液的配制:
38.分别精密称取维生素a(上海源叶hplc≥96%)、维生素a醛(上海源叶)、维生素a酸(上海源叶hplc≥99%)、维生素a醋酸酯(上海源叶hplc≥99%)、维生素a棕榈酸酯(上海源叶hplc≥96%)20mg,置于5ml的容量瓶中,加入适量的抗坏血酸无水乙醇使其溶解,β-胡萝卜素(上海源叶hplc≥98%)20mg,置于5ml的容量瓶中,加入适量的抗坏血酸三氯甲烷溶液使其溶解,分别定容至5ml,配制成浓度为4mg/ml的标准储备液。
39.(2)混合标准溶液的配制
40.分别精密吸取6种维生素a族化合物标准储备液125μl置于10ml容量瓶中,用抗坏血酸乙醇溶液稀释、定容,配制成浓度为100μg/ml的混合标准储备液。将混合标准储备液进行逐级稀释,分别配制成浓度为50μg/ml、25μg/ml、10μg/ml、5μg/ml、2.5μg/ml、1μg/ml、0.5μg/ml的混合标准工作液。
41.(3)样品溶液的制备
42.皂化样品前处理过程为,称取约10.0g的匀浆样品(精确至0.1g),向其中加入等量抗坏血酸和5ml(50g/100g)氢氧化钾溶液,将15ml无水乙醇溶液分三次加入匀浆样品中进行60℃超声(600w)提取30min,将提取液合并,分次加入20ml乙酸乙酯萃取三次,将萃取液合后用蒸馏水水洗至萃取液经ph试纸检测呈中性,经5g无水硫酸钠干燥后,将萃取液在40℃条件下减压蒸发,待剩下1-2ml液体时停止旋转蒸发,取下旋蒸瓶用氮气吹干,用5ml抗坏血酸乙醇三氯甲烷混合溶液溶解后,用有机膜过滤后进高效液相色谱仪进行分析。
43.非皂化样品前处理过程为,称取约10.0g的匀浆样品(精确至0.1g),向其中加入等量抗坏血酸,将15ml无水乙醇溶液分三次加入匀浆样品中进行60℃超声(600w)提取30min,将提取液合并,分次加入20ml乙酸乙酯萃取三次,将萃取液合后用蒸馏水水洗至萃取液经ph试纸检测呈中性,经5g无水硫酸钠干燥后,将萃取液在40℃条件下减压蒸发,待剩下1-2ml液体时停止旋转蒸发,取下旋蒸瓶用氮气吹干,用5ml抗坏血酸乙醇三氯甲烷混合溶液溶解后,用有机膜过滤后进高效液相色谱仪进行分析。
44.(4)标准曲线的绘制
45.分别取7个不同浓度的混合标准工作液,从高浓度到低浓度依次进高效液相色谱仪进行定量测定,色谱条件为采用ymc-βcarotene c
30
(4.6mm
×
250mm,3μm)色谱柱,柱温为30℃、进样量为20μl、流速为0.8ml/min、检测波长为325nm、350nm、380nm、460nm,梯度洗脱程序为:0-8min 90%a(甲醇)、1%b(超纯水)、9%c(甲基叔丁基醚);8.1min35%a(甲醇)、65%c(甲基叔丁基醚);8.1-16min 35%a(甲醇)、65%c(甲基叔丁基醚);16.1min 90%a(甲醇)、1%b(超纯水)、9%c(甲基叔丁基醚)。以浓度(c)为横坐标,对应峰面积(a)为纵坐标进行标准曲线的绘制。
46.表1线性回归方程及方法的检测限、定量限和线性范围
[0047][0048]
检测结果如表1所示,6种维生素a族化合物的检测浓度范围在0.5-50μg/ml范围内,其浓度与峰面积呈现良好的线性关系。
[0049]
实施例2:
[0050]
与实施例1中步骤(1)~(4)基本相同,仅做如下改变,色谱条件为:采用ymc-βcarotene c
30
(4.6mmx250 mm,3μm)色谱柱,柱温为30℃、进样量为20μl、流速为0.8ml/min、检测波长为325nm、350nm、380nm、460nm,梯度洗脱程序为:0-8min 90%a(乙腈)、1%b(超纯水)、9%c(甲基叔丁基醚);8.1min 35%a(乙腈)、65%c(甲基叔丁基醚);8.1-16min 35%a(乙腈)、65%c(甲基叔丁基醚);16.1min 90%a(乙腈)、1%b(超纯水)、9%c(甲基叔丁基醚)。
[0051]
检测结果如图2所示,乙腈、超纯水、甲基叔丁基醚三种流动相在固定的梯度洗脱程序下,维生素a、全反式维生素a醛、维生素a醋酸酯的出峰时间均在8min,三者分离度低,维生素a和全反式维生素a醛出峰时间完全重合,β-胡萝卜素峰型对称性差。
[0052]
实施例3:
[0053]
与实施例1中步骤(1)~(4)基本相同,仅做如下改变,色谱条件为:采用ymc-βcarotene c
30
(4.6mm
×
250mm,3μm)色谱柱,柱温为30℃、进样量为20μl、流速为0.8ml/min、检测波长为325nm、350nm、380nm、460nm,梯度洗脱程序为:0-8min 90%a(乙腈10%甲醇)、1%b(0.01%磷酸溶液)、9%c(甲基叔丁基醚);8.1min 35%a(乙腈10%甲醇)、65%c(甲基叔丁基醚);8.1-16min 35%a(乙腈10%甲醇)、65%c(甲基叔丁基醚);16.1min 90%a(甲醇)、1%b(0.01%磷酸溶液)、9%c(甲基叔丁基醚)。
[0054]
检测结果如图3所示,维生素a酸和全反式维生素a醛分离效果较差,其余4种维生素a族化合物峰型较为清楚、分离效果良好。
[0055]
实施例4:
[0056]
与实施例1中步骤(1)~(4)基本相同,仅做如下改变,色谱条件为:采用ymc-βcarotene c
30
(4.6mmx250 mm,3μm)色谱柱,柱温为30℃、进样量为20μl、流速为1.0ml/min、检测波长为325nm、350nm、380nm、460nm,梯度洗脱程序为:0-8min 90%a(甲醇)、1%b(0.01%磷酸溶液)、9%c(甲基叔丁基醚);8.1min 35%a(甲醇)、65%c(甲基叔丁基醚);8.1-16min 35%a(甲醇)、65%c(甲基叔丁基醚);16.1min 90%a(甲醇)、1%b(0.01%磷酸溶液)、9%c(甲基叔丁基醚)。
[0057]
检测结果如图4所示,全反式维生素a醛的检测出峰效果差且峰型不对称,说明了进一步增大甲醇的比例(无乙腈)对维生素a酸和全反式维生素a醛的峰型、对称性等不产生影响。
[0058]
实施例5:精密度试验
[0059]
取实施事例1中的浓度为25μg/ml的混合标准工作溶液,按照实施例1步骤(4)的液相色谱方法,进行液相检测,连续测定6次,记录峰面积。
[0060]
表2精密度试验结果
[0061]
[0062][0063]
从表2精密度试验结果来看,6种维生素a族化合物峰面积的rsd值均小于1%(n=6),表明该方法具有良好的精密度。
[0064]
实施例6:重复性试验
[0065]
取同一批次的厚壳贻贝样品,按照实施例1步骤(3)中所示方法制备样品溶液,按照实施事例1步骤(4)的液相色谱方法,进行液相检测,记录峰面积。
[0066]
表3重复性试验结果
[0067][0068]
如表3所示,厚壳贻贝样品经非皂化前处理后,含有维生素a棕榈酸酯和β-胡萝卜素,经皂化处理后,含有维生素a棕榈酸酯和β-胡萝卜素,且rsd均小于3%,表明该方法重复性良好。
[0069]
实施例7:稳定性试验
[0070]
按照实施例1步骤(3)中所示方法制备厚壳贻贝样品溶液,分别在室温下放置0、3、6、9、12、24h后,按照实施事例1步骤(4)的液相色谱方法,进行液相检测,记录峰面积。
[0071]
表4稳定性试验结果
[0072][0073]
如表4所示,厚壳贻贝样品经非皂化前处理后,含有维生素a棕榈酸酯和β-胡萝卜素,经皂化处理后,含有维生素a棕榈酸酯和β-胡萝卜素,且rsd均小于2%,表明该方法稳定性良好。
[0074]
实施例8:加标回收试验
[0075]
将厚壳贻贝样品进行分别皂化和非皂化前处理,然后进行维生素a族化合物的分析测定,同时进行3个平行样检测,将测定浓度结果作为本底值。然后向3个同一样品里分别
加入20μg/ml、10μg/ml、5μg/ml三个高中低不同浓度的维生素a族化合物混合标准品溶液后按照样品前处理步骤分别进行处理,而后在确定的液相色谱条件下进行高效液相色谱分析,每个浓度进行3个平行样分析检测。计算加标回收率。
[0076]
表5非皂化法加标回收试验结果
[0077][0078]
表6皂化法加标回收实验结果
[0079][0080]
如表5和表6所示,非皂化法维生素a的加标回收率为87.32%(平均值),维生素a醋酸酯的加标回收率为96.82%(平均值),维生素a棕榈酸酯的加标回收率为90.81%(平均值),维生素a酸的加标回收率为112.65%(平均值),全反式维生素a醛的加标回收率为57.39%(平均值),β-胡萝卜素的加标回收率为111.01%(平均值),且六种维生素a类化合物的rsd均小于2%,表明样品测定较准确。皂化法维生素a的加标回收率为85.4%(平均值),β-胡萝卜素的加标回收率为97.65%(平均值),且这两种维生素a类化合物的rsd均小于2%,表明样品测定较准确。
[0081]
实施例9:准确性试验
[0082]
配制浓度为40μg/ml的6种维生素a族化合物的混合标准品溶液,按照实施事例1步骤(4)的方法,进行液相检测,计算其实际浓度。结果表明,维生素a、维生素a醋酸酯、维生素a棕榈酸酯、维生素a酸、全反式维生素a醛、β-胡萝卜素的测定浓度分别为39.85μg/ml、40.10μg/ml、40.26μg/ml、40.66μg/ml、38.53μg/ml、39.03μg/ml,与实际浓度接近,因而,该方法具有良好的准确度。
[0083]
实施例10:实际样品的测定
[0084]
分别选取厚壳贻贝、牡蛎、扇贝、毛蚶、缢蛏5种贝类样品,按照实施例1中步骤(3)制备样品溶液,按照实施事例1步骤(4)的液相色谱方法,进行液相检测,记录样品中目标物的峰面积,通过标准曲线计算其含量。
[0085]
表7非皂化法贝类维生素a族化合物测定结果
[0086][0087]
表8皂化法贝类维生素a族化合物测定结果
[0088][0089][0090]
如上表所示,非皂化法处理后,厚壳贻贝和扇贝中只检测到了维生素a棕榈酸酯和β-胡萝卜素,牡蛎中检测到了维生素a、维生素a棕榈酸酯和β-胡萝卜素,毛蚶和缢蛏中只检
测到了β-胡萝卜素。皂化法处理后,在厚壳贻贝、牡蛎、扇贝中检测到了维生素a和β-胡萝卜素,毛蚶和缢蛏中只检测到了β-胡萝卜素。且毛蚶中β-胡萝卜素含量较高。
[0091]
本发明所述实施例为本实验优选的实施方式,并不限于上述实施方式,在不背离本发明的实质内容的情况下,本领域技术人员能够做出的任何显而易见的改进、替换或变型均属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1