一种检测地美硝唑的分子印迹电化学传感器及其制备方法
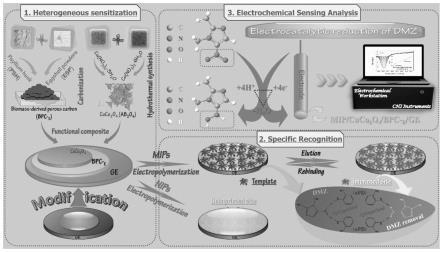
1.本发明属于食品安全领域,尤其是涉及一种检测地美硝唑的分子印迹电化学传感器及其制备方法。
背景技术:
2.地美硝唑(dmz)是一种兽药,被广泛应用于治疗由细菌和原虫感染引起的疾病,不仅如此,dmz还起到了提高饲料效率和诱导动物生长的促进作用。然而,dmz引起的残留不仅会对人类健康造成危害,还会影响生态系统的平衡。直接接触过量的dmz会导致皮肤发炎、视力丧失、呼吸道刺激、肺损伤、癌症、不孕不育和肾衰竭等恶性疾病;即使在痕量水平下,dmz也能引起不良的副作用,包括脑病和周围神经性病变。因此,研发高效、简便、灵敏、快速和良好选择性的分析方法来检测dmz浓度水平是至关重要的。dmz作为一个电活性分子,可在未修饰的电极表面直接发生电还原反应,其缓慢的表面动力学,严重影响了检测dmz的灵敏度,而存在的共存物其电还原峰电位也会与dmz的还原电位发生重叠,使传感器失去了实际检测的应用价值。
3.由于生物质衍生多孔碳纳米材料的前体具有选择多样性、巨大可获得性、快速再生和环境友好等优势已在电化学传感领域引起了广泛的关注。引入的蛋壳(es)粉末在原位碳化制备生物质(圆苞车前子壳,psh)衍生碳纳米材料过程中不仅可显著提高其合成材料的物化性能,如增大比表面积、孔径、孔容等,而且还提高了生物质衍生多孔碳材料的表面缺陷密度,进而增加了修饰电极电化学反应的活性位点数量以及对后修饰物的结合位点。具有尖晶石结构的cuco2o4纳米材料,由于二元金属间的协同作用,具备了优异的电化学活性,由于cuco2o4纳米材料性能不仅取决于其组成,还在一定程度上受其微观结构特征的影响。为此也探究了不同形貌、不同粒径、不同孔径分布的cuco2o4纳米材料。将上述两种纳米材料经电极表面层层组装设计,开发一种优异的电极敏化纳米材料也具有重要的研究意义和价值。
4.为有效提高电化学传感器的选择性,仿生人工受体“分子印迹聚合物(mips)”,相较于酶、抗体等生物活性分子,是一种拥有广阔前景的识别材料,其高度交联的多孔mips具有与目标分子形状和大小互补的特定识别位点,对目标分子表现出了卓越的选择性。此外,构建在电极修饰剂表面的mips可充分暴露识别位点,从而提高扩散率、传质率、结合能力和位点可及性。
技术实现要素:
5.有鉴于此,本发明旨在克服现有技术中的缺陷,提出一种检测地美硝唑的分子印迹电化学传感器及其制备方法。
6.为达到上述目的,本发明的技术方案是这样实现的:
7.一种检测地美硝唑的分子印迹电化学传感器的制备方法,包括如下步骤:
8.(1)将ge电极的表面进行清理、抛光、冲洗后得到处理后的ge电极;
9.(2)将生物质衍生多孔碳分散于溶剂中,得到均匀的悬浊液,将该悬浊液与粘合剂溶液混合后滴涂在处理后的ge电极表面,待溶剂挥发后,得到bpc-e
/ge;
10.(3)将铜基二元金属氧化物配置成悬浊液,将该悬浊液滴涂在所述的bpc-e
/ge电极表面,烘干后得到cuco2o4/bpc-e
/ge;
11.(4)将所述的cuco2o4/bpc-e
/ge浸没在含有邻苯二胺与地美硝唑的缓冲溶液中,进行电聚合,然后进行清洗、吹干,得到mip/cuco2o4/bpc-e
/ge前体;
12.(5)将所述的mip/cuco2o4/bpc-e
/ge前体经磁力搅拌洗脱,使模板分子地美硝唑从所述的mip/cuco2o4/bpc-e
/ge前体中去除,得到所述的mip/cuco2o4/bpc-e
/ge印迹电极。
13.进一步,所述的步骤(2)中的悬浊液的浓度为1-5mg
·
ml-1
;所述的步骤(2)中的粘合剂溶液为0.05-0.5%的壳聚糖-醋酸溶液;所述的步骤(2)中的悬浊液与粘合剂溶液的体积比为20-5:1;所述的步骤(2)中的溶剂为乙醇溶液;所述的乙醇溶液中乙醇与水的体积比为1:1;所述的步骤(2)中的滴涂步骤的滴涂量为10μl。
14.进一步,所述的步骤(3)中的悬浊液为cuco2o4乙醇-nafion悬浊液;所述的步骤(3)中的悬浊液的浓度为1-10mg
·
ml-1
;所述的步骤(3)中的滴涂步骤的滴涂量为10μl。
15.进一步,所述的步骤(4)中的缓冲溶液为甲醇-醋酸缓冲溶液;所述的步骤(4)中的缓冲溶液中邻苯二胺(opd)的浓度为0.5-5mm,地美硝唑的浓度为0.1-0.5mm;所述的步骤(4)中的电聚合步骤的电位为0-0.8v,扫描速率为50-100mv
·
s-1
;所述的电聚合步骤的电聚合圈数为5-30圈;所述的步骤(5)中的洗脱步骤的洗脱液为甲醇-乙酸溶液,其中甲醇与乙酸的体积比为10-6:1,洗脱时间为5-20min。
16.一种电极敏化纳米材料生物质衍生多孔碳的制备方法,包括如下步骤:将圆苞车前子壳粉和蛋壳粉末进行干燥、混合后焙烧,将得到的剩余物研磨后置于溶液中磁力搅拌、冲洗、离心、再次干燥后得到所述的电极敏化纳米材料生物质衍生多孔碳。bpc-e
具有高密度表面缺陷的介孔结构特性。
17.进一步,所述的焙烧步骤的升温速率为1-10℃
·
min-1
,从室温加热至600-900℃,并保温0.5-8h;所述的干燥步骤的温度为100-150℃,时间为8-24h;所述的再次干燥步骤的温度为50-100℃,时间为8-24h。
18.一种铜基二元金属氧化物的制备方法,包括如下步骤:
19.(1)将无机金属铜盐和无机金属钴盐溶于去离子水中,得到混合溶液a;
20.(2)将co(nh2)2和nh4f混合在去离子水中,得到溶液b;
21.(3)将溶液b在磁搅拌作用下缓慢滴入溶液a中,将得到的混合物水热合成后得到所述的铜基二元金属氧化物。得到的cuco2o4为形貌规整、尺寸均匀的立方形纳米材料。
22.进一步,所述的步骤(1)中的无机金属铜盐为cu(no3)2·
3h2o、cucl2或cuso4中的至少一种;所述的步骤(1)中的无机金属钴盐为co(no3)2·
6h2o、cocl2、coso4、cof2或coco3中的至少一种;所述的步骤(1)中的无机金属铜盐和无机金属钴盐中cu
2+
和co
2+
的摩尔比为1:2;所述的步骤(3)中的水热合成步骤的温度为100-200℃,保温4-10h。
23.所述的电极敏化纳米材料生物质衍生多孔碳的用途,所述的电极敏化纳米材料生物质衍生多孔碳在检测地美硝唑中的应用。
24.所述的铜基二元金属氧化物的用途,所述的铜基二元金属氧化物在检测地美硝唑中的应用。
25.相对于现有技术,本发明具有以下优势:
26.本发明所述的检测地美硝唑的分子印迹电化学传感器基于电极敏化纳米材料和靶向识别mips两种材料的优势,首先在ge表面层层组装敏化纳米材料(bpc-e
和cuco2o4),修饰的电极cuco2o4/bpc-e
/ge增加了电极界面的电子传输能力,以此为基底在其表面电聚合mips,经洗脱液洗脱,制备出的印迹电极mip/cuco2o4/bpc-e
/ge具有多重靶向识别位点和催化位点,对dmz分子有良好的选择识别性和电催化效果。
27.本发明所述的检测地美硝唑的分子印迹电化学传感器首次利用psh和es粉末两种生物质制备了物化性能优良的bpc-e
,由于原位引入的es粉末具有模板和活化双重效应,其作为电极修饰材料可提高电子传输及后修饰材料负载的能力;在bpc-e
/ge表面组装了形貌可控、尺寸均匀的介孔cuco2o4纳米材料,借助两种异质材料的协同效应,cuco2o4/bpc-e
/ge的敏化性能明显增强,进一步加速了电子和质量的传递过程;在此基础上,引入的mips膜又赋予了修饰电极的靶向识别能力;在最优条件下,所构建的分子印迹电化学传感器对dmz具有灵敏度高的检测性能,在浓度范围0.01-10μm和10-200μm分别呈现良好的线性关系,其检出限为1.8nm(s/n=3),灵敏度为5.724μa
·
μm-1
·
cm-2
。与已有的分析方法相比,该传感器具备了高灵敏、强特异性和重复性、重现性、稳定性好等优点。
附图说明
28.构成本发明创造的一部分的附图用来提供对本发明创造的进一步理解,本发明创造的示意性实施例及其说明用于解释本发明创造,并不构成对本发明创造的不当限定。在附图中:
29.图1为本发明实施例3和4所述的传感材料的示意图。
30.图2为本发明实施例3所述的bpc-e
扫描电镜图(a为500nm尺度下的扫描电镜图、b和c分别为100nm尺度下不同区域的扫描电镜图、a、b和c分别为bpc-e
中c、n和o元素的分布图);cuco2o4纳米材料的扫描电镜图(d为2μm尺度下的扫描电镜图、e和f分别为500nm尺度下不同区域的扫描电镜图、d、e和f分别为cuco2o4中cu、co和o元素的分布图)。
31.图3为本发明实施例3所述的bpc-e
的透射电镜图(a、c、d、e和f分别为100、50、10和5nm尺度下不同区域的透射电镜图)、b为bpc-e
的选区电子衍射图(saed);cuco2o4纳米材料的透射电镜图(g、i、j、k和l分别为100、20、10和5nm尺度下不同区域的透射电镜图)、h为cuco2o4的选区电子衍射图(saed)。
32.图4为本发明实施例4所述的电聚合mips的循环伏安曲线(cvs)图。
33.图5为本发明实施例4所述的洗脱时间优化图。
34.图6为本发明实施例5所述的印迹电极和非印迹电极洗脱前后及印迹电极对dmz再吸附的循环伏安曲线(cvs)图。
35.图7为本发明实施例6所述的dmz电催化对比线性扫描伏安曲线(lsvs)图。
36.图8为本发明实施例7所述的不同浓度dmz的差分脉冲伏安曲线(dpvs)图和标准曲线图。
37.图9为本发明实施例7所述的选择性实验图。
具体实施方式
38.除有定义外,以下实施例中所用的技术术语具有与本发明所属领域技术人员普遍理解的相同含义。以下实施例中所用的试验试剂,如无特殊说明,均为常规生化试剂;所述实验方法,如无特殊说明,均为常规方法。
39.下面结合实施例来详细说明本发明。
40.实施例1
41.一种电极敏化纳米材料生物质衍生多孔碳(bpc-e
)和铜基二元金属氧化物(cuco2o4)的制备方法:
42.圆苞车前子壳(psh)粉和蛋壳(es)粉末在使用前先在105℃下干燥24h。称取3g psh和3g es粉末,混合后放入管式炉中,设定升温速率为2℃
·
min-1
,从室温加热至600℃,并恒温保持0.5h。将得到的剩余物充分研磨,在2m hcl溶液中磁力搅拌12h,反复冲洗离心,直至上清液ph为7,收集剩余物并在60℃真空烘箱中干燥24h。
43.将1.2g cu(no3)2·
3h2o和2.9g co(no3)2·
6h2o前体溶于20ml去离子水中,得到混合溶液a;将1.2g co(nh2)2和0.18g nh4f混合在10ml去离子水中,得到溶液b。之后,将溶液b在磁搅拌作用下缓慢滴入溶液a中,并加入0.5ml氨水,得到的混合物转移至100ml晶化釜内,100℃恒温保持4h。
44.实施例2
45.圆苞车前子壳(psh)粉和蛋壳(es)粉末在使用前先在105℃下干燥24h。称取3g psh和3g es粉末,混合后放入管式炉中,设定升温速率为10℃
·
min-1
,从室温加热至900℃,并恒温保持8h。将得到的剩余物充分研磨,在2m hcl溶液中磁力搅拌12h,反复冲洗离心,直至上清液ph为7,收集剩余物并在60℃真空烘箱中干燥24h。
46.将1.2g cu(no3)2·
3h2o和2.9g co(no3)2·
6h2o前体溶于20ml去离子水中,得到混合溶液a;将1.2g co(nh2)2和0.18g nh4f混合在10ml去离子水中,得到溶液b。之后,将溶液b在磁搅拌作用下缓慢滴入溶液a中,并加入5ml氨水,得到的混合物转移至100ml晶化釜内,200℃恒温保持10h。
47.实施例3
48.圆苞车前子壳(psh)粉和蛋壳(es)粉末在使用前先在105℃下干燥24h。称取3g psh和3g es粉末,混合后放入管式炉中,设定升温速率为5℃
·
min-1
,从室温加热至850℃,并恒温保持2h。将得到的剩余物充分研磨,在2m hcl溶液中磁力搅拌12h,反复冲洗离心,直至上清液ph为7,收集剩余物并在60℃真空烘箱中干燥24h。
49.将1.2g cu(no3)2·
3h2o和2.9g co(no3)2·
6h2o溶于20ml去离子水中,得到混合溶液a;将1.2g co(nh2)2和0.18g nh4f混合在10ml去离子水中,得到溶液b。之后,将溶液b在磁搅拌作用下缓慢滴入溶液a中,并加入1ml氨水,得到的混合物转移至100ml晶化釜内,200℃恒温保持8h。
50.为了更好地理解本发明制备的bpc-e
和cuco2o4纳米材料,对其进行了扫描电镜、透射电镜和元素分布的表征。
51.从图2a至2c中可以看出制备的bpc-e
表面结构粗糙,可以在表面暴露更多的边、角、台,从而提供更多的吸附/活性位点。图2a至2c说明了bpc-e
的主要元素由碳、氧和少量的氮元素组成,与psh的原料组成元素一致,确定了制备的bpc-e
的纯度和组成。图3(a、c和d)展示
了bpc-e
存在波纹形态的纳米片结构,该结构不仅提供了连续的电子路径以保证良好的导电性,而且其扩散途径促进了传质过程。saed的衍射环和弥散晕斑(见图3b)证实了bpc-e
多晶和非晶结构的存在,说明了sp
2-c和缺陷共存在bpc-e
纳米材料的表面。同时,进一步观察发现(如图3e和3f),bpc-e
存在不完全晶格条纹,进一步证明了其表面存在大量的缺陷。
52.从图2d可以看出合成的cuco2o4具有尺寸均匀的立方型形貌,放大倍数后(见图2e和2f),其表面呈现粗糙崎岖的形态,这样的结构可以大大提供更多的电活性位点,从而促进电化学反应的进行。元素分布图(图2d、e和f)可以看出cu、co和o元素均呈现良好的分布。图3k和3l展示了cuco2o4纳米材料的晶格间距为2.87和依次对应于cuco2o4的(220)和(311)位面。图3a至3d可以观察到cuco2o4纳米材料存在间隙缺陷、层错、位错和孪晶界等表面缺陷。图3h所测量的saed呈现5个衍射环,依次与cuco2o4的(220)、(311)、(400)、(511)和(440)位面相对应,说明具有多晶性质的cuco2o4拥有独立的随机生长方向。
53.实施例4
54.(1)cuco2o4/bpc-e
/ge制备
55.将ge电极表面滴涂食人鱼溶液5min后,依次用不同粒径(1.0、0.3和0.05μm)的氧化铝浆抛光,再分别用无水乙醇和去离子水冲洗,得到干净的ge。bpc-e
超声分散在乙醇-水中(v:v=1:1),得到浓度为2mg
·
ml-1
的悬浊液,并按体积比为10:1的方式与0.1%壳聚糖-醋酸溶液进行混合,取上述溶液10μl滴涂在ge表面,待溶剂挥发后,得到bpc-e
/ge。随后,配制浓度为5mg
·
ml-1
cuco2o4乙醇-nafion悬浊液,量取10μl滴涂在bpc-e
/ge电极表面,红外灯下烘干得到cuco2o4/bpc-e
/ge。
56.(2)mip/cuco2o4/bpc-e
/ge制备
57.预先配制含1.5mm的邻苯二胺(opd)单体和0.3mm dmz的甲醇-醋酸缓冲溶液(0.2m ph=5.2),之后cuco2o4/bpc-e
/ge浸没在上述溶液中,在窗口电位0到0.8v,扫描速率100mv
·
s-1
条件下电聚合10圈,并用去离子水清洗,氮气吹干,得到mip/cuco2o4/bpc-e
/ge前体,以甲醇-乙酸(v:v=9:1)为洗脱液,经磁力搅拌洗脱10min,使模板分子dmz从mip/cuco2o4/bpc-e
/ge前体中去除,得到印迹电极mip/cuco2o4/bpc-e
/ge。
58.如图4所示,随着聚合圈数的增加,电流值呈现减小的趋势,说明mips前体能被良好的聚合到cuco2o4/bpc-e
/ge表面。之后如图5所示,以甲醇-乙酸(v:v=9:1)为洗脱液,mip/cuco2o4/bpc-e
/ge前体经洗脱后,在10min时获得了最大的响应电流值,这是由于洗脱出的dmz分子使印迹电极表面形成了与dmz相匹配的印迹空腔,促进了探针溶液的电子传输能力,致使电流响应值增加。
59.实施例5
60.图6研究了印迹电极(mip/cuco2o4/bpc-e
/ge)和非印迹电极(nip/cuco2o4/bpc-e
/ge)洗脱前后的循环伏安曲线(cvs)及印迹电极对dmz的再吸附情况。洗脱前,印迹电极(a)和非印迹电极(b)在探针溶液中均未出现氧化还原峰电流,说明mips和nips修饰膜良好地覆盖在cuco2o4/bpc-e
/ge表面,抑制了电子转移过程。经甲醇-乙酸(v:v=9:1)溶液洗脱后,印迹电极(c)氧化还原峰电流更明显,而对应的非印迹电极几乎没有变化,说明dmz模板分子被成功洗脱,随后产生与dmz互补的印迹空腔,而非印迹电极(d)没有任何空腔的形成,这是由于未添加模板分子dmz。重新吸附100μm dmz后,印迹电极的氧化还原峰电流(e)明显降低,表明dmz分子重新嵌入空洞所致。
61.实施例6
62.由于dmz是一种电化学活性物质,它可以在给定的电位下发生还原反应。如图7所示,考察了在0.04m b-r缓冲液(ph=6.8)中孵育100μm dmz不同修饰电极的电化学行为。印迹电极表现出最大的还原峰电流密度,这是由于印迹电极的印迹空腔预先富集溶液中的dmz,使dmz在电极表面的浓度升高,进而提高了dmz的电化学反应效率,上述结果也进一步证明了所制备的印迹电极具有良好的识别、富集和电催化能力。
63.实施例7
64.图8为不同浓度dmz的差分脉冲曲线(dpvs),随着dmz浓度的升高,其还原峰电流值(-i
pc
)呈上升趋势,在浓度范围0.01-10μm和10-200μm中,拟合的线性方程为-i
pc
(μa)=0.916c
dmz
(μm)+6.403(r2=0.9943)和-i
pc
(μa)=0.117c
dmz
(μm)+14.075(r2=0.9935),所得检出限为1.8nm(s/n=3)灵敏度为5.724μa
·
μm-1
·
cm-2
。结果说明制备的传感器对dmz测定具有宽的检测浓度范围、低的检出限和高的灵敏度。
65.选择2倍于100μm dmz的干扰物质,包括奥硝唑(onz)、甲硝唑(mnz)、磺胺醋酰(sa)、磺胺胍(sgn)、尿酸(ua)、青霉胺(dpa)、罗硝唑(rnz)、氨丙啉(amr)、替硝唑(tnz)作为研究该电化学器的选择性。如图9所示,引入的干扰物质对dmz测定无影响,说明构建的电化学器具有良好的选择性。
66.以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1