一种同时测定肉类中多种β-受体激动剂的液质联用检测方法
一种同时测定肉类中多种
β-受体激动剂的液质联用检测方法
技术领域
1.本发明涉及食品检测领域,具体涉及一种测定肉类中β-受体激动剂的检测方法。
背景技术:
2.肉类因其含有丰富的营养价值,一直是人类饮食的基本组成部分,随着生活水平的提高,肉类消费量呈现逐年增长的趋势。近年来人们的健康观念由高脂肪、高能量食物的摄入转向消费低脂、低糖的食物改变,这也给传统的畜禽养殖带来了改变,如瘦肉型的生猪更受到人们的青睐。
3.β-受体激动剂又称β-兴奋剂,是一类具有肾上腺素功能的苯乙醇胺类人工合成化合物,起初被开发是用于治疗人或动物的子宫、呼吸系统和心血管疾病。后来研究人员发现当β-受体激动剂的使用剂量超过治疗剂量5-10倍时,可以减少脂肪沉积、促进蛋白质合成、显著提高胴体的瘦肉率和饲料转换率。因此,不法商家开始在动物饲料中添加β-受体激动剂以谋取非法利益。
4.β-受体激动剂化学结构稳定,常常在动物体内难以降解,容易经过食物链进一步在人体内残留,当人体积累到一定量时会导致严重的不良反应,如肌肉振颤、头晕、心跳加速或急性中毒,甚至直接导致死亡。
5.为了保护消费者的健康,许多国家或组织对β-受体激动剂的使用做出了相关规定,如欧盟立法禁止所有用于动物促生长作用的β-受体激动剂,同时限定了各类动物组织中的最大残留限量(mrls)。中国也明文禁止在饲料和动物饮用水中使用此类药物,并将β-受体激动剂类药物作为国家农产品质量安全监督抽查和例行监测的物质。美国、加拿大、日本等国家规定了残留标准,但没有禁止使用所有β-受体激动剂。由此可见,制定科学准确的β-受体激动剂检测方法不仅保障动物源性食品安全,也是应对国际贸易壁垒的有效措施。
6.目前饲料及动物性食品中对β-受体激动剂类药物的分析方法主要有免疫筛选方法和仪器确证方法。免疫筛选方法包括酶联免疫法(enzyme-linked immunosorbent assay,elisa)和胶体金免疫层析法(gold immunochromatographyassay,gica)。仪器分析方法有毛细管电泳法(capillary electrophoresis,ce)、气相色谱-串联质谱法(gas chromatography-mass spectrometry,gc-ms)、液相/超高效液相色谱-串联质谱法(liquid phase/ultra high performance liquid chromatography-mass spectrometry,lc/uplc-ms/ms)等。此外,近年来又有拉曼、传感器和生物芯片法等方法。其中免疫筛选方法存在抗原抗体制备困难、检测灵敏度不高、易出现假阳性结果的问题,常常只用于市场的日常筛选检测,传感器等新型的检测方法应用较少,有待进一步研究。仪器确证方法中的uplc-ms/ms法属于lc-ms/ms法,因其分析时间更短、灵敏度和准确度更高的优点,已成为目前主流定性和定量手段,其检测过程一般包括酶解、有机溶剂提取、固相萃取净化、上机测定等操作,然而该方法具有前处理操作较为繁琐费时、有机溶剂消耗大、检测种类较少等缺点。
7.为有效控制动物源性食品中β-受体激动剂的残留问题,目前亟待发明一种前处理快速简单、能同时准确测定多种β-受体激动剂的分析检测方法。
技术实现要素:
8.为解决上述技术问题,本发明包括以下几个方面:
9.本发明的第一方面提供一种同时测定肉类中多种β-受体激动剂的液质联用分析检测方法,所述方法包括以下步骤:
10.(1)酶解:称取均质后的肉类,加入乙酸铵和水解酶,混匀后避光酶解,离心取上清液;
11.(2)提取:在步骤(1)的上清液中加入有机溶剂,提取后加入nacl沉淀蛋白,涡旋后离心,取上清液;
12.(3)纯化:取步骤(2)的上清液过一步法萃取柱,将收集的滤液用氮气吹干,用复溶液复溶,充分混匀后过滤,得待测样品;
13.(4)测定:使用超高效液相色谱-串联质谱仪对步骤(3)的待测样品进行检测;其中色谱条件为:色谱柱为反相色谱柱,流动相为水-有机溶剂二元溶剂体系,洗脱方式为梯度洗脱程序。
14.优选的,所述步骤(1)中的肉类为猪肉、牛肉或羊肉。
15.优选的,所述步骤(1)中的肉类酶解前加入100μg/l的内标工作液。
16.优选的,所述步骤(1)中的乙酸铵的浓度为0.2mol/l。
17.优选的,所述步骤(1)中的水解酶为β-葡萄糖醛苷酶/芳基硫酸酯酶。
18.优选的,所述步骤(1)中的酶解条件是40℃下酶解2h。
19.优选的,所述步骤(1)中的离心条件是10000r/min高速离心机中在4℃下离心5min。
20.优选的,所述步骤(2)中的有机溶剂是含1%乙酸的乙腈。
21.优选的,所述步骤(2)中的离心条件是4℃下10000r/min离心5min。
22.优选的,所述步骤(3)中的萃取柱选自oasis mcx柱、oasis hlb柱、bond elut c18柱或者qvet-ag柱。
23.更优选的,所述步骤(3)中的萃取柱是qvet-ag柱。
24.优选的,所述步骤(3)中的复溶液是体积比为90:10的0.1%甲酸水:甲醇溶液。
25.优选的,所述步骤(3)中的过滤是通过0.22μm ptfe有机相滤膜。
26.最优选的,所述方法包括如下步骤:准确称取均质后的猪、牛、羊肌肉组织2.00
±
0.02g,置于50ml离心管中,加入浓度为100μg/l的混合内标工作液50μl、0.2mol/l的乙酸铵10ml和40μlβ-葡萄糖醛苷酶/芳基硫酸酯酶,涡旋混匀1min,置于40℃下避光水酶解2h,酶解后冷却至室温,涡旋1min后于10000r/min高速离心机中在4℃下离心5min,取上清液置于另一洁净50ml离心管中,加入10ml含1%乙酸的乙腈提取,并加入2gnacl沉淀蛋白,涡旋5min后,4℃下10000r/min离心5min,取上清液2ml过qvet-ag一步法萃取柱净化,以1滴/秒的速度直接收集滤液,40℃下水浴氮气吹干,1ml 0.1%甲酸水:甲醇为90:10的复溶液复溶,充分混匀1min后过0.22μm ptfe有机相滤膜,收集于进样瓶中,供超高效液相色谱-串联质谱仪检测。
27.优选的,所述步骤(4)中的色谱柱选自acquity uplc hss c18柱、thermo scientific hypersil gold柱或者thermo scientific accucoretm aq柱。
28.更优选的,所述色谱柱为thermo scientific hypersil gold柱。
29.优选的,所述步骤(4)中的流动相选自水-甲醇、水-乙腈、0.1%甲酸水-0.1%甲酸甲醇或者0.1%甲酸水-0.1%甲酸乙腈。
30.更优选的,所述流动相为0.1%甲酸水-0.1%甲酸甲醇。
31.优选的,所述步骤(4)中的梯度洗脱程序选自:
32.梯度洗脱程序1:
33.时间a%b%0964296412406012.196416964
34.梯度洗脱程序2:
35.时间a%b%095567030960401110901295516955
36.或者梯度洗脱程序3:
37.时间a%b%0955295557030960401210901395516955
38.,其中a相为水相,b相为有机相。
39.更优选的,所述梯度洗脱程序为梯度洗脱程序3:
40.时间a%b%0955295557030960401210901395516955
41.,其中a相为水相,b相为有机相。
42.进一步优选的,所述步骤(4)中的色谱条件如下:色谱柱为thermo scientific hypersil gold柱;流动相a为0.1%甲酸水,流动相b为0.1%甲酸甲醇;流速为0.3ml/min;柱温30℃;3μl进样;梯度洗脱程序为:
43.时间(min)流动相a(%)流动相b(%)0955295557030960401210901395516955。
44.优选的,所述步骤(4)中的质谱条件如下:电喷雾电离正离子模式(esi
+
);多反应监测模式(mrm);喷雾电压(is):3500v;离子源温度(tem):450℃;离子传输管温度(ion transfer tube temp):300℃;雾化器温度(vaporizer temp):280℃;鞘气流速(sheath gas):38arb;辅助气流速(aux gas):4arb。
45.优选的,16种β-受体激动剂及5种内标物的质谱采集参数如下:
[0046][0047][0048]
,其中带*的为定量子离子。
[0049]
优选的,所述β-受体激动剂选自(1)克伦特罗、(2)马布特罗、(3)溴布特罗、(4)克伦潘特、(5)莱克多巴胺、(6)沙丁胺醇、(7)特布他林、(8)福莫特罗、(9)异克舒令、(10)利托
君、(11)苯乙醇胺a、(12)氯丙那林、(13)妥布特罗、(14)班布特罗、(15)西布特罗和(16)羟甲基克伦特罗中的2种以上。
[0050]
更优选的,所述β-受体激动剂选自上述16种化合物中的3种以上、4种以上、5种以上、6种以上、7种以上、8种以上、9种以上、10种以上、11种以上、12种以上、13种以上、14种以上或者15种以上。
[0051]
进一步优选的,所述β-受体激动剂为以下16种化合物的组合物:(1)克伦特罗、(2)马布特罗、(3)溴布特罗、(4)克伦潘特、(5)莱克多巴胺、(6)沙丁胺醇、(7)特布他林、(8)福莫特罗、(9)异克舒令、(10)利托君、(11)苯乙醇胺a、(12)氯丙那林、(13)妥布特罗、(14)班布特罗、(15)西布特罗和(16)羟甲基克伦特罗。
[0052]
本发明的第二方面提供色谱条件在改善同时测定肉类中多种β-受体激动剂的液质联用分析检测方法中的应用,所述色谱条件为(1)色谱柱为thermo scientific hypersil gold柱;和(2)流动相a为0.1%甲酸水,流动相b为0.1%甲酸甲醇,梯度洗脱程序为:
[0053]
时间(min)流动相a(%)流动相b(%)0955295557030960401210901395516955。
[0054]
优选的,上述应用中所述β-受体激动剂为以下16种化合物的组合物:(1)克伦特罗、(2)马布特罗、(3)溴布特罗、(4)克伦潘特、(5)莱克多巴胺、(6)沙丁胺醇、(7)特布他林、(8)福莫特罗、(9)异克舒令、(10)利托君、(11)苯乙醇胺a、(12)氯丙那林、(13)妥布特罗、(14)班布特罗、(15)西布特罗和(16)羟甲基克伦特罗。
[0055]
本发明的第三方面提供前处理条件在改善同时测定肉类中多种β-受体激动剂的液质联用分析检测方法中的应用,所述前处理条件为(1)在待测肉类中加入β-葡萄糖醛苷酶/芳基硫酸酯酶进行酶解;(2)酶解液中加入含1%乙酸的乙腈提取;(3)提取液过qvet-ag柱萃取;和(4)萃取液挥干后使用体积比为90:10的0.1%甲酸水:甲醇溶液作为复溶液复溶。
[0056]
优选的,上述应用中所述β-受体激动剂为以下16种化合物的组合物:(1)克伦特罗、(2)马布特罗、(3)溴布特罗、(4)克伦潘特、(5)莱克多巴胺、(6)沙丁胺醇、(7)特布他林、(8)福莫特罗、(9)异克舒令、(10)利托君、(11)苯乙醇胺a、(12)氯丙那林、(13)妥布特罗、(14)班布特罗、(15)西布特罗和(16)羟甲基克伦特罗。
[0057]
本发明产生的技术效果:
[0058]
1、本发明建立了畜肉中检出率高的16种β-受体激动剂的高效液相色谱-串联质谱检测方法,在样品前处理过程中优化了酶解条件,大大缩短了检测的时间,对样品进行提取后,采用一步法qvet-ag固相萃取柱净化,提高了检测效率的同时减少了有机试剂的消耗,
整体前处理时长不超过6小时。
[0059]
2、本发明建立的高效液相色谱-串联质谱检测方法通过优化色谱和质谱条件,在16min内即可检测完毕,16种化合物在0.25-125μg/kg范围内线性良好,检测方法灵敏度、重复性好、分析时间短且检测化合物数量多,适合于动物源性食品中β-受体激动剂的日常检测与确证分析,有助于对市场中β-受体激动剂类化合物兽药残留的监管。
附图说明
[0060]
图1是使用thermo scientific accucoretm aq柱分离16种β-受体激动剂及5种内标物的总离子流图;
[0061]
图2是使用thermo scientific accucoretm aq柱(a)和thermo scientific hypersil gold柱(b)分离氯丙那林的色谱峰对比图;
[0062]
图3是使用thermo scientific accucoretm aq柱(a)和thermo scientific hypersil gold柱(b)分离克伦特罗的色谱峰对比图;
[0063]
图4是使用thermo scientific accucoretm aq柱(a)和thermo scientific hypersil gold柱(b)分离特布他林的色谱峰对比图;
[0064]
图5是使用acquity uplc hss c18柱分离16种β-受体激动剂及5种内标物的总离子流图;
[0065]
图6是使用acquityuplc hss c18柱(a)和thermo scientific hypersil gold柱(b)分离西布特罗的色谱峰对比图;
[0066]
图7是使用acquityuplc hss c18柱(a)和thermo scientific hypersil gold柱(b)分离特布他林的色谱峰对比图;
[0067]
图8是使用acquityuplc hss c18柱(a)和thermo scientific hypersil gold柱(b)分离福莫特罗的色谱峰对比图;
[0068]
图9是使用水-乙腈作为流动相分离16种β-受体激动剂及5种内标物的总离子流图;
[0069]
图10是使用水-乙腈(a)与0.1%甲酸水-0.1%甲酸甲醇(b)作为流动相分离氯丙那林的色谱峰对比图;
[0070]
图11是使用水-乙腈(a)与0.1%甲酸水-0.1%甲酸甲醇(b)作为流动相分离苯乙醇胺a的色谱峰对比图;
[0071]
图12是使用0.1%甲酸水-0.1%甲酸乙腈作为流动相分离16种β-受体激动剂及5种内标物的总离子流图;
[0072]
图13是使用0.1%甲酸水-0.1%甲酸乙腈(a)与0.1%甲酸水-0.1%甲酸甲醇(b)作为流动相分离沙丁胺醇的色谱峰对比图;
[0073]
图14是使用0.1%甲酸水-0.1%甲酸乙腈(a)与0.1%甲酸水-0.1%甲酸甲醇(b)作为流动相分离特布他林的色谱峰对比图;
[0074]
图15是使用0.1%甲酸水-0.1%甲酸乙腈(a)与0.1%甲酸水-0.1%甲酸甲醇(b)作为流动相分离西布特罗的色谱峰对比图;
[0075]
图16是使用水-甲醇作为流动相分离16种β-受体激动剂及5种内标物的总离子流图;
8、p-9、p-10、b-1、b-2、b-3、b-4、b-5、b-6、b-7、b-8、b-9、b-10、l-1、l-2、l-3、l-4、l-5、l-6、l-7、l-8、l-9、l-10)市售生鲜畜肉来自中国三个不同的省份,编号p、b、l分别代表猪肉、牛肉、羊肉,数字1-4采自江苏省,5-7采自黑龙江省,8-10采自广西省。采集的畜肉样品经hm6300均质机(中国莱谱科技有限公司)在10000r/min条件下均质,-20℃条件下保存。
[0094]
1.3、标准溶液的配制
[0095]
100mg/l的16种标准品及5种内标作为储备液保存在-20℃冰箱中,有效期12个月,各取100μl单标于10ml容量瓶中,用甲醇定容至刻度,得到1mg/l混合标准中间溶液,于4℃冰箱中保存,有效期1个月;分别移取100μl内标单标储备液,用甲醇溶解并定容至10ml容量瓶中,配制成1mg/l内标混合标准溶液,于4℃冰箱中保存,有效期1个月;根据实验需要,用甲醇稀释1mg/l内标混合标准溶液成100μg/l内标混合标准工作液,现用现配。
[0096]
1.4、试验仪器及检测条件
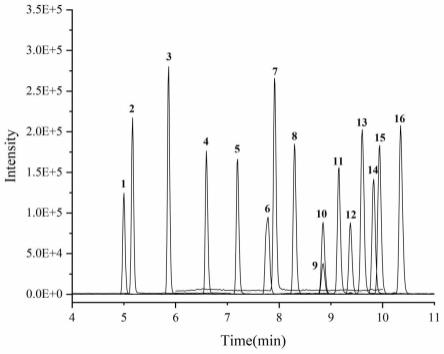
[0097]
使用vanquish&tsq quantis(配有xcalibur工作站软件,版本号4.1.31.9,美国thermo fisher scientific公司)超高效液相色谱质谱仪进行检测。如无特别说明,本试验例的液质联用检测条件如下:
[0098]
色谱条件:色谱柱为thermo scientific hypersil gold柱;流动相a为0.1%甲酸水,流动相b为0.1%甲酸甲醇;流速为0.3ml/min;柱温30℃;3μl进样;梯度洗脱程序为:
[0099]
时间(min)流动相a(%)流动相b(%)0955295557030960401210901395516955。
[0100]
质谱条件:电喷雾电离正离子模式(esi
+
);多反应监测模式(mrm);喷雾电压(is):3500v;离子源温度(tem):450℃;离子传输管温度(ion transfer tube temp):300℃;雾化器温度(vaporizer temp):280℃;鞘气流速(sheath gas):38arb;辅助气流速(aux gas):4arb。
[0101]
1.5、样品制备
[0102]
如无特别说明,本试验例的样品按照下述方法制备:准确称取(xs105电子天平,瑞士mettler-toledo公司)均质后的猪、牛、羊肌肉组织(2.00
±
0.02g),置于50ml离心管中,加入50μl混合内标工作液(浓度为100μg/l)、0.2mol/l乙酸铵(乙酸调ph为5.2)10ml和40μlβ-葡萄糖醛苷酶/芳基硫酸酯酶,涡旋(vortex genius涡旋混匀器,德国ika公司)混匀1min,置于40℃下避光水(tw20,德国julabo)酶解2h,酶解后冷却至室温,涡旋1min后于10000r/min d-16c高速离心机(德国sartorious公司)中在4℃下离心5min,取上清液置于另一洁净50ml离心管中,加入10ml 1%乙酸乙腈提取,并加入2g nacl沉淀蛋白,涡旋5min后,4℃下10000r/min离心5min,取上清液2ml过qvet-ag(6ml,2g,日本shimadzu公司)一步法萃取柱净化,以1滴/秒的速度直接收集滤液,40℃下水浴氮气(n-evap型氮吹仪,美国
organomation公司)吹干,1ml复溶液(0.1%甲酸水:甲醇=90:10)复溶,充分混匀1min后过0.22μm ptfe有机相滤膜(美国pall公司)收集于进样瓶中,供超高效液相色谱-串联质谱仪检测。
[0103]
1.6、数据分析
[0104]
使用tracefinder 4.1软件(美国thermo fisher scientific公司)进行数据采集。使用excel 2016软件(美国microsoft公司)进行数据处理。使用origin 2022b软件(美国originlab公司)绘图。
[0105]
2、检测条件的优化
[0106]
2.1、色谱柱的优化
[0107]
16种β-受体激动剂为中等或强极性化合物,目前大部分文献都采用反相色谱柱进行分离,本试验例比较了acquityuplc hss c18柱(2.1mm
×
150mm,1.7μm,美国waters公司)、thermo scientific hypersil gold(100mm
×
2.1mm,1.9μm,美国thermo fisher scientific公司)和thermo scientific accucoretm aq柱(2.1mm
×
150mm,2.6μm,美国thermo fisher scientific公司)对β-受体激动剂的分离效果,其他色谱条件如下:流动相为0.1%甲酸水-甲醇,洗脱程序为梯度洗脱程序2,进样量为3μl,流速为0.3ml/min,柱温为30℃。
[0108]
试验结果表明,thermo scientificaccucoretm aq柱分离氯丙那林、克伦特罗时出现拖尾峰、分离特布他林时出现前沿峰,而thermo scientific hypersil gold分离上述化合物的色谱峰的峰形较好(参见图1-4)。acquityuplc hss c18柱分离西布特罗出现分叉峰,分离特布他林出现前沿峰,分离福莫特罗时出现响应低,基线高的问题,而thermo scientific hypersil gold分离上述化合物的色谱峰峰型较好(参见图5-8)。综上,thermo scientific hypersil gold对16种β-受体激动剂的分离效果最好,同时缩短了分析时间,故选择thermo scientific hypersil gold作为优选的色谱柱。
[0109]
2.2、流动相的优化
[0110]
本试验例分别比较水-甲醇、水-乙腈、0.1%甲酸水-0.1%甲酸甲醇、0.1%甲酸水-0.1%甲酸乙腈四种流动相对16种β-受体激动剂进行高效液相色谱分离的分离效果,其他色谱条件如下:色谱柱为thermo hypersil gold柱,洗脱程序为梯度洗脱程序2,进样量为3μl,流速为0.3ml/min,柱温为30℃。
[0111]
试验结果表明,使用水-乙腈作为流动相分离氯丙那林、苯乙醇胺a时存在基线噪声大,杂峰多的问题,而使用0.1%甲酸水-0.1%甲酸甲醇分离上述化合物时无明显杂峰(参见图9-11);使用0.1%甲酸水-0.1%甲酸乙腈作为流动相分离沙丁胺醇、特布他林时存在前沿峰,分离西布特罗时存在分叉峰,而使用0.1%甲酸水-0.1%甲酸甲醇分离上述化合物时色谱峰的峰形较好(参见图12-15);使用水-甲醇作为流动相分离妥布特罗、马布特罗、异克舒令时存在响应低,基线噪声大的问题,而使用0.1%甲酸水-0.1%甲酸甲醇分离上述化合物时基线噪音较小(参见图16-19)。综上所述,选择0.1%甲酸水-0.1%甲酸甲醇作为流动相时,可以使得极性相近的β-受体激动剂被分段洗脱出,且与溶剂保持一致,可以获得尖锐对称的峰形,信号响应高,因此流动相组成优选0.1%甲酸水-0.1%甲酸甲醇。
[0112]
2.3、梯度洗脱条件的优化
[0113]
因为16种β-受体激动剂极性相近,等度洗脱难以实现这16种物质的分离,因此选
择梯度洗脱方式。为确定优选的梯度洗脱程序,本试验例设计了以下3种梯度洗脱强度:
[0114]
梯度洗脱程序1:
[0115]
时间a%b%流速(ml/min)09640.329640.31240600.312.19640.3169640.3
[0116]
梯度洗脱程序2:
[0117]
时间a%b%流速(ml/min)09550.3670300.3960400.31110900.3129550.3169550.3
[0118]
梯度洗脱程序3:
[0119]
时间a%b%流速(ml/min)09550.329550.3570300.3960400.31210900.3139550.3169550.3
[0120]
其他色谱条件如下:色谱柱选择thermo scientific hypersil gold,流动相a为0.1%甲酸水;流动相b为0.1%甲酸甲醇。
[0121]
试验结果表明,使用梯度洗脱程序1和2分离特布他林和沙丁胺醇时均存在分叉峰的问题,而使用梯度洗脱程序3分离上述化合物时色谱峰的峰形较好(参见图20-23),因此最终确定优选梯度洗脱程序3,分析时间为16min。综上,当色谱柱选择thermo scientific hypersil gold柱,流动相选择0.1%甲酸水-0.1%甲酸甲醇,梯度洗脱选择梯度洗脱程序3时,16种化合物分离效果最好(参见图24,加标浓度为10μg/kg;色谱峰含义如下:1:teb;2:sal;3:cib;4:rit;5:h-clb;6:rac;7:clo;8:clb;9:pea;10:fom;11:tul;12:brm;13:mab;14:clp;15:iss;16:bam;虽然pea和fom在色谱中同时出现色谱峰,但是它们可以进一步通过质谱分离)。
[0122]
2.4、质谱条件的优化
[0123]
将100μg/l的16种单标工作液和5种单个内标工作液分别采用针泵进样方式以3μl/min流速注入质谱,在正离子模式下开启q1全扫描,确定每种标准物的分子离子,然后以
此作为母离子,进行子离子扫描,选取丰度较强、干扰较小的子离子为定性离子。在多反应监测(mrm)模式下优化各种质谱参数,优化得到各种化合物的质谱参数见表1。
[0124]
表116种β-受体激动剂及5种内标物的质谱采集参数
[0125][0126]
[0127]
注:带*的为定量子离子
[0128]
2.5、样品前处理方法的优化
[0129]
2.5.1、酶解条件的优化
[0130]
苯酚型β-受体激动剂的轭合代谢作用强,易生成各种轭合物,一般采用酸水解和酶解等方式使结合态的β-受体激动剂转变成游离态,便于生物体内残留量的准确定量,也多采用葡萄糖醛酸酶进行酶解,但酶解的温度和时间对于酶解作用效果具有显著影响。基于此,确定采用β-葡萄糖醛苷酶/芳基硫酸酯酶,根据比较不同的温度和时间对酶解效果的影响,以空白加标回收率作为指标,试验结果表明(见表2),40℃下酶解2h的效果与37℃下酶解12h的效果相当,而55℃下酶解时,异克舒令、妥布特罗、莱克多巴胺等7种化合物的回收率不佳,可能温度影响酶的活性进而影响了药物的回收。因此,本试验例酶解条件优选在40℃下酶解2h,与gb/t 21313-2007、gb/t 22286-2008、sn/t1924-2011采用37℃避光酶解16h相比,有效缩短了实验室前处理时间,提高了检测效率。
[0131]
表2酶解条件对16种β-受体激动剂回收率的影响
[0132][0133]
2.5.2、提取溶剂的优化
[0134]
β-受体激动剂是在苯乙醇胺母核的基础上进行官能团修饰和改造后形成的一类化合物,具有水溶性强和中等极性或强极性的特点,此类化合物残留分析一般采用甲醇、乙腈作为提取试剂,可以降低高脂肪、高蛋白的基质干扰,本试验例比较不提取、甲醇和1%乙酸乙腈提取对目标化合物加标回收率的影响。
[0135]
试验结果表明,不提取直接净化基质干扰严重,多种化合物回收率不佳,与甲醇相比,乙腈提取时带入的极性干扰物质更少,后续净化效果更好;而在乙腈中加入少量的酸,更有利于弱碱性的β-受体激动剂提取(见表3)。因此,本试验例提取溶剂优选含1%乙酸的乙腈。
[0136]
表3提取溶剂对16种β-受体激动剂回收率的影响
[0137][0138]
2.5.3、固相萃取柱的优化
[0139]
由于动物源性食品中基质成分复杂,即使经过含1%乙酸的乙腈提取后,提取液中仍然有很多杂质干扰目标物在仪器上的响应,需要进一步的净化处理。固相萃取技术(spe)是近年来快速发展的样品前处理技术,能够纯化富集目标物,相比传统的液液萃取,具有溶剂用量少、操作方便、稳定性好等优点。因此,本试验例采用固相萃取柱对提取液进一步富集净化,并比较了oasis mcx柱(60mg/3ml,美国waters公司)、oasis hlb柱(200mg/3ml,美
国waters公司)、bond elut c18柱(200mg/3ml,美国agilent公司)和qvet-ag等四种固相萃取柱对16种β-受体激动剂的净化效果,以16种化合物的加标回收率作为依据,试验结果显示,qvet-ag柱对样品提取液的净化效果最好,所有化合物加标回收率满足要求(见表4)。另外,与常规的萃取柱相比,qvet-ag柱无需进行活化、平衡、淋洗、洗脱等步骤,操作步骤更简便的同时也减少了有机试剂的消耗,体现了简单环保的理念,因此本试验例的纯化方法优选qvet-ag萃取小柱。
[0140]
表4纯化方法对16种β-受体激动剂回收率的影响
[0141][0142]
2.5.4、复溶液的优化
[0143]
样品上机前复溶液的确定直接影响方法的灵敏度,本试验例将10μg/l混合标准溶液氮气吹干,采用不同有机相比例(0.1%甲酸水:甲醇体积比分别为90:10、70:30、50:50)的复溶液复溶,发现加入一定比例的有机相使目标物更易于溶解,明显提高了16种化合物的响应,三种复溶液均能满足回收率的要求,但使用有机相比例过大时易产生溶剂效应,沙丁胺醇、特布他林峰形较差(参见图25),可能会影响定量结果的准确性,因此最终选择0.1%甲酸水:甲醇体积比为90:10作为复溶液。
[0144]
3、方法验证
[0145]
3.1、基质效应评估
[0146]
基质效应(matrix effect,me)是指样品中其他组分对目标化合物分析过程产生的干扰,从而影响定量结果的准确性。本发明采用空白猪肉、牛肉、羊肉基质样品,按前述确定的样品处理方法得到空白基质液配制的标准曲线(化合物浓度梯度为0.1、0.5、1.0、2.0、5.0、10.0、50.0μg/l),并与复溶液(体积比0.1%甲酸水:甲醇=90:10)稀释得到的溶剂标准曲线进行比较,通过计算基质标准曲线与试剂标准曲线斜率比值的关系(见计算公式1)评估基质效应。
[0147]
me(%)=ka/kb
×
100 (1)
[0148]
ka为基质匹配标准曲线的斜率,kb为溶剂标准曲线的斜率;当me>1时,为基质增强效应;me<1时为基质抑制效应;me=1时则不存在基质干扰。
[0149]
从图26可知,3种基质对大部分化合物为基质抑制效应,抑制率大小不同,不同的基质对同一种化合物的基质效应也不相同;由于食品基质的复杂性以及对目标物痕量分析的要求,本发明方法采用基质匹配标准曲线法及同位素内标法定量,不仅可以减少前处理所引入的误差,而且能够补偿基质效应对定量分析结果的影响。
[0150]
3.2、方法线性范围、检出限与定量限
[0151]
基于已建立的uplc-ms/ms方法,将配制好的基质匹配标准工作溶液在优化的仪器条件下上机测定,以各化合物定量离子色谱峰面积与相应内标的峰面积之比为纵坐标(y),对应化合物浓度(μg/l)与内标物浓度之比为横坐标(x)进行线性拟合,绘制标准曲线,得到
回归方程并计算对应的平方相关系数(square ofcorrelation coefficient,r2),以信噪比(s/n)≥3时所对应的加标浓度作为方法检出限(lod)、s/n≥10时的加标浓度作为定量限(loq)。
[0152]
试验结果显示,16种β-受体激动剂在0.1-50μg/l(相当于样品中0.25-125μg/kg)浓度范围内线性良好,相关系数≥0.9928,根据信噪比确定方法lod为0.01-0.11μg/kg,loq为0.04-0.38μg/kg(参见表5),这一结果低于gb/t 22286-2008中所规定的lod(0.5μg/kg),进一步说明该方法灵敏度高。
[0153]
表5畜肉中16种β-受体激动的线性方程、检出限和定量限
[0154]
[0155][0156]
3.3、加标回收率与精密度试验
[0157]
因β-受体激动剂在中国属于禁用物质,在确定样品预处理方法后,根据gb/t27404-2008《实验室控制规范食品理化检测》中附录f-检测方法确认的技术要求评价回收率和精密度(见计算公式2和3),对于食品中禁用物质,回收率应该以方法测定低限、2倍方法测定低限、10倍方法测定低限进行三水平实验。每个浓度水平进行6次平行测试,共重复3批次,测量结果的相对标准偏差(rsd)作为日内和日间精密度,以此评价检测方法稳定性。
[0158]
回收率(%)=加标试样测定量/实际加标量
×
100(2)
[0159]
精密度(%)=标准偏差/平均值
×
100(3)
[0160]
试验结果表明,三种畜肉样品基质中的空白加标回收实验结果表明所有化合物的加标回收率和精密度结果均满足要求,不同添加水平下猪肉、牛肉和羊肉平均回收率范围分别为62.62%-115.93%、61.35%-106.34%、62.00%-111.83%,所有化合物在三种基质中的批内、批间精密度(rsd)分别为0.66%-9.99%、2.12%-9.98%,符合回收率范围满足60-120%、精密度小于20%的要求,说明该方法测得畜肉中16种β-受体激动剂具有较好的准确度,方法稳定性良好。
[0161]
3.4、实际样品应用
[0162]
为了验证方法的有效性,随机选取来自中国不同省份的猪肉、牛肉、羊肉样品各10份(共30份),按本发明确定的方法检测其中的16种β-受体激动剂残留量,检测过程中各类药物通过空白添加的方式进行质控,进一步验证该方法的可行性与准确度。
[0163]
在阳性样品检测过程中发现,来自江苏省的一份牛肉样品检出克伦特罗值为2.454μg/kg,检出率为3.33%,检出项目克伦特罗的质谱图和色谱图如图27所示。检测过程中通过空白加标的克伦特罗牛肉质控样加标量5μg/kg,测定值为4.677μg/kg,回收率良好,阳性样品的检出及质控样的验证结果进一步证明了该方法的可行性及准确性。
[0164]
综上所述,本发明建立的分析检测方法灵敏度、重复性好、分析时间短且检测化合物数量多,适合于动物源性食品中β-受体激动剂的日常检测与确证分析,有助于对市场中β-受体激动剂类化合物兽药残留的监管。
[0165]
虽然已经对本发明的具体实施方案进行了描述,但是本领域技术人员应认识到,在不偏离本发明的范围或精神的前提下可以对本发明进行多种改变与修饰。因而,本发明意欲涵盖落在附属权利要求书及其同等物范围内的所有这些改变与修饰。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1