一种氟化钪中Sc含量的检测方法与流程
一种氟化钪中sc含量的检测方法
技术领域
1.本发明涉及sc含量的检测方法,特别是氟化钪中sc含量的检测方法。
背景技术:
2.公开号为cn114563394a的中国专利中公开了一种氟化钪中杂质含量的检测方法,包括如下步骤:(1)称取适量的氟化钪作为试样,将氟化钪用氢氧化钠、碳酸钠和碳酸氢钠混合熔融,冷却后加水形成混合溶液;(2)将盐酸、硼酸加入混合溶液中,得到澄清溶液;(3)用icp-oes检测澄清溶液中不同杂质的光谱强度,根据标准曲线计算得到氟化钪中杂质含量。该专利能准确测量氟化钪中杂质((mg、bi、ba、v、se)的含量,但是采用滴定法测量钪含量时,滴定时终点无法达到亮黄色,无法准确测量氟化钪中sc含量。
技术实现要素:
3.本发明所要解决的技术问题是,针对现有技术不足,提供一种氟化钪中sc含量的检测方法,滴定时终点可以达到亮黄色,可以准确测量氟化钪中sc含量。
4.为解决上述技术问题,本发明所采用的技术方案是:一种氟化钪中sc含量的检测方法,包括以下步骤:
5.s1、将氟化钪与强碱溶液充分混匀加热,使氢氧化钪沉淀完全;
6.s2、过滤氢氧化钪沉淀,弃去滤液,用水洗涤氢氧化钪沉淀直至水洗液中无氟离子存在;
7.s3、在s2水洗之后的氢氧化钪沉淀中,加入盐酸,加热溶解,盐酸和氢氧化钪反应生成氯化钪溶液;
8.s4、冷却至室温后,转入容量瓶中,混匀;
9.s5、分取试样,调节ph为1.5-1.8,滴加二甲酚橙指示剂,用edta-2na标准溶液滴定至亮黄色,根据消耗的edta-2na标准溶液的量,计算得到氟化钪中钪含量。
10.发明人在反复研究中意外发现,现有方法氟离子与钪离子都在溶液中,氟离子跟溶液中钪离子络合,抑制钪离子与edta-2na反应,钪含量检测结果偏低,且滴定终点颜色突变不明显。
11.本发明使氟化钪转为氢氧化钪沉淀,通过洗涤氢氧化钪沉淀使钪与氟分离,消除氟离子对钪含量测定的干扰,使用edta-2na标准滴定溶液直接检测钪的含量,检测效率高,准确性高。
12.在本发明的一个优选的实施例中,所述强碱溶液为氢氧化钠,强碱溶液可使氟化钪得到充分的沉淀。
13.在本发明的一个优选的实施例中,氢氧化钠的浓度为8%-15%。本技术中严格控制氢氧化钠的浓度为8%-15%,当氢氧化钠的浓度低于8%时,后续检测结果的标准偏差较大,当氢氧化钠的浓度高于15%时,过滤时滤纸被烫卷,无法过滤而无法实现钪离子与氟离子的分离。
14.在本发明的一个优选的实施例中,氟化钪与氢氧化钠的质量比为0.25:24-45。反应过程中氢氧化钪的添加量高于氟化钪与氢氧化钠的摩尔比3:1,氢氧化钠需过量,保证充分生成氢氧化钪。
15.在本发明的一个优选的实施例中,s1中加热的温度为160-200℃,加热过程中持续搅拌40-60min,使氟化钪与氢氧化钠处于溶液(不能蒸干)状态下反应。
16.本技术中溶解时间严格控制在40-60min,时间过短,钪转换成氢氧化钪沉淀不完全,检测结果偏低;溶解时间过长,溶液中氢氧化钠的浓度过高,过滤时滤纸被烫卷,无法过滤而无法实现钪离子与氟离子的分离。
17.在本发明的一个优选的实施例中,s5中用盐酸或氨水调节溶液调节ph。
18.在本发明的一个优选的实施例中,s3中加热的温度为200-240℃,加热时间为30-50min。
19.在本发明的一个优选的实施例中,s3中盐酸的加入量为每0.25g氟化钪,s3中加入20-30ml盐酸。
20.优选的,s3中加入盐酸,使得s3中酸度大于1.5,充分溶解氢氧化钪沉淀。
21.优选的,s5中加入2滴二甲酚橙指示剂。
22.优选的,锌标准溶液(1mg/mlzn)的配置方法:称取0.2g(精确至0.0001g)纯锌(质量分数﹥99.9%)于250烧杯中,加10ml水、缓慢分次加入10ml盐酸,低温加热至完全溶解。溶液移入200ml容量瓶中,加5ml盐酸,以水稀释至刻度,混匀。
23.优选的,二甲酚橙(2g/l)指示剂的配置方法:称取0.2g二甲酚橙,溶于水,稀释至100ml,混匀。
24.优选的,乙酸—乙酸钠缓冲液(ph5.5—6)的配置方法:称取结晶乙酸钠200克溶于适量的水中,加入10ml冰乙酸,用水稀至1000ml,混匀。
25.优选的,edta-2na标准溶液的配置方法为:称取37.2克edta溶于1l热水中,用水稀释至10l,混匀。
26.优选的,edta-2na标准溶液的标定方法:取10ml锌标准溶液(1mg/ml zn)于250ml烧杯中,加2滴甲基橙指示剂,滴加(1+1)氨水中和,使溶液由橙色刚显黄色,以少量水冲洗杯壁,加入乙酸-乙酸钠缓冲溶液20ml,加二甲酚橙指示剂1滴,用edta-2na标准液滴至由酒红色转变成亮黄色为终点。
27.公开号为cn114563394a的中国专利存在以下缺陷:
28.1、现有方法用氢氧化钠、碳酸钠、碳酸氢钠混合熔融,钪及氟均存在于溶液中,过滤无法将钪及氟进行分离。
29.2、现有方法氟离子跟钪离子均在溶液中,现有方法加入硼酸络合氟离子,使其不干扰icp-oes对杂质元素的测定;但该处理方式无法消除溶液中的氟对edta-2na滴定钪的测定干扰。
30.氟化钪作为高纯金属钪生产的重要中间产物,氟化钪中钪含量影响着高纯金属钪的质量,准确检测氟化钪中钪含量尤为重要。氟化钪的氟离子干扰edta-2na滴定钪元素的检测,消除氟化钪中氟离子的干扰,是准确检测氟化钪中钪含量的必要条件。
31.1、本发明主要解决的问题是将钪离子与氟离子分离,消除氟离子对edta-2na滴定钪的终点判定(滴定终点颜色为亮黄色,见【图4】);2、现有方法氟离子跟钪离子均在溶液
中,通过加入硼酸使氟离子不干扰icp-oes对杂质元素的测定;氟离子干扰edta-2na滴定钪的终点判定(滴定时终点无法达到亮黄色,见【图3】)。
32.所述氟化钪中钪含量浓度值按以下进行计算:
[0033][0034]
式中:
[0035]
c——edta-2na标准溶液的浓度,单位为摩尔每升(mol/l);
[0036]
m——钪的摩尔质量44.96,单位为克每摩尔(g/mol);
[0037]
v——滴定试液消耗edta-2na标准溶液的体积,单位为毫升(ml);
[0038]
v1——试液的总体积,单位为毫升(ml);
[0039]
v2——分取试液的体积,单位为毫升(ml);
[0040]
m0——称取试料的质量,单位为克(g)。
[0041]
与现有技术相比,本发明所具有的有益效果为:
[0042]
本发明先将氟化钪转为氢氧化钪沉淀,通过洗涤氢氧化钪沉淀使钪与氟分离,消除氟离子对钪含量测定的干扰,使用edta-2na标准滴定溶液直接检测钪的含量,溶解时间短,使用试剂少,检测效率高,准确性高、成本低。
[0043]
本发明进行了精密度以及加标回收率实验,结果表明本发明的检测方法操作方便,结果准确,检测结果稳定性、准确性符合相关要求。此外,本发明的检测方法工艺合理,安全可靠,操作简便,费用较低,满足生产需要。
附图说明
[0044]
图1为本发明一实施例的检测流程图。
[0045]
图2为现有方法的检测流程图。
[0046]
图3为本发明一实施例中钪与氟离子未分离,氟离子干扰edta-2na滴定钪的终点判定。
[0047]
图4为现有方法中钪与氟离子分离后,edta-2na滴定钪的终点判定。
具体实施方式
[0048]
下面对本发明的具体实施方式作进一步说明。在此需要说明的是,对于这些实施方式的说明用于帮助理解本发明,但并不构成对本发明的限定。此外,下面所描述的本发明各个实施方式中所涉及的技术特征只要彼此之间未构成冲突就可以相互组合。
[0049]
下述实施例中的实验方法,如无特殊说明,均为常规方法,下述实施例中所用的试验材料,如无特殊说明,均为可通过常规的商业途径购买得到的。
[0050]
实施例1
[0051]
1.主要仪器设备:电热板、磁力搅拌器
[0052]
2.试剂:盐酸(gr)、氢氧化钠(gr)、edta-2na(ar)、氨水(ar)、二甲酚橙(ar)、冰醋酸(ar)、醋酸钠(ar)、纯锌(基准试剂)。
[0053]
3.试验过程
[0054]
按照图1所示,检测氟化钪中钪含量:
[0055]
3.1准确称取0.25g(精确至0.0001g)氟化钪试样于250ml聚四氟烧杯中,与氢氧化钠溶液(10%)300ml充分混匀,其中氢氧化钠的质量为30g;
[0056]
3.2设置180℃加热搅拌60min,形成氢氧化钪沉淀;
[0057]
3.3过滤氢氧化钪沉淀,氟离子在滤液中弃去,继续用水洗涤沉淀直至滤液中无氟离子存在;
[0058]
3.4将沉淀转入原烧杯中,加30ml盐酸,盐酸的浓度为36-38%;
[0059]
3.5于电热板上设置200℃加热40min,将氢氧化钪转为氯化钪溶液。
[0060]
3.6冷却至室温后,转入100ml容量瓶中,混匀;
[0061]
3.7分取5.00ml试样,加水至70ml,用盐酸或氨水(当ph值酸度小于1.5时,用氨水调节;当ph值酸度大于1.8时,用盐酸调节)调节溶液ph为1.5-1.8,加入2滴二甲酚橙指示剂,用edta-2na标准溶液滴定至亮黄色,根据消耗的edta-2na标准溶液的量,计算得到氟化钪中钪含量。
[0062]
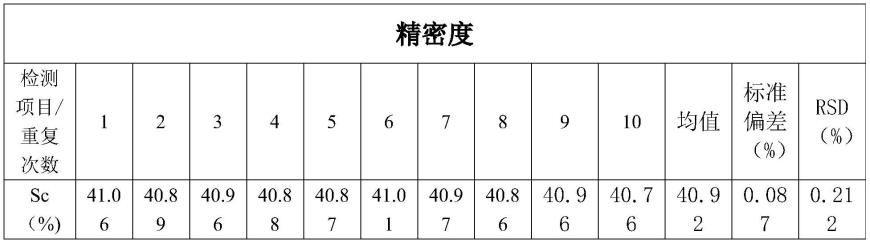
3.8本发明检测方法的精密度
[0063]
称取样品1份,按照本发明检测方法测试,结果如下:
[0064]
表1同一试样平行测定10次的检测结果和精密度
[0065][0066]
标准偏差sd的计算公式:
[0067][0068]
相对标准偏差rsd的计算公式:
[0069][0070]
按照本发明检测样品的精密度符合gb/t27404-2008中的表f.2(实验室内变异系数)。
[0071]
3.9本发明的回收率
[0072]
选取1份氟化钪样品,按照本发明检测方法处理后,测定结果,通过计算,称取一定重量的试样和相应的钪标准,记为标称值,进行回收率实验,结果如下:
[0073]
表2实施例1的回收率
[0074]
[0075][0076]
上表表明,采用本发明的检测方法测定氟化钪中钪含量的方法回收率达83.50至93.00%,符合国家标准gb/t27404-2008《实验室质量控制规范食品理化检测》中对回收率试验的规定。
[0077]
对比例1
[0078]
1.一种氟化钪中钪含量测试试剂包括:盐酸(ar)、氢氧化钠(ar)、碳酸钠(ar)、碳酸氢钠(ar)、硼酸(ar)、edta-2na(ar)、氨水(ar)、二甲酚橙(ar)、冰醋酸(ar)、醋酸钠(ar)、纯锌(基准试剂)、去离子水。
[0079]
一种氟化钪中钪含量的检测方法,包括以下步骤:
[0080]
2.准确称取0.1g(精确至0.0001g)氟化钪试样于250ml聚四氟烧杯中
[0081]
3.加入0.5g(精确至0.01g)氢氧化钠、0.3g(精确至0.01g)碳酸钠、0.3g(精确至0.01g)碳酸氢钠
[0082]
4.向上述试剂中加入10ml去离子水,轻轻混匀
[0083]
5.将上述混合物置于电炉上加热消解,于180-220℃加热蒸干
[0084]
6.取下冷却至室温后加入30ml盐酸,180-220℃加热30-60min
[0085]
7.冷却至室温后,加入40g/l硼酸溶液5-10ml,混匀后放置30min
[0086]
8.用去离子水将溶液定容至100ml摇匀,冷却至室温,溶液为澄清的水溶液。
[0087]
9.分取5.00ml试样,加水至70ml,用盐酸或氨水调节溶液ph为1.5-1.8,加入2滴二甲酚橙指示剂,用edta-2na标准溶液滴定至亮黄色,根据消耗的edta-2na标准溶液的量,计算得到氟化钪中钪含量。
[0088]
10.现有方法的精密度
[0089]
称取样品1份,按照现有方法测试,结果如下:
[0090][0091][0092]
标准偏差sd的计算公式:
[0093]
[0094]
相对标准偏差rsd的计算公式:
[0095][0096]
按照现有方法检测氟化钪样品中钪含量的精密度不符合gb/t27404-2008中的表f.2(实验室内变异系数)。
[0097]
11.现有方法的回收率
[0098]
选取1份氟化钪样品,按照现有方法检测方法处理后,测定结果,通过计算,称取一定重量的试样和钪标准,记为标称值,进行回收率实验,结果如下:
[0099]
表4对比例1的回收率
[0100][0101]
由上表可知,采用现有方法测定氟化钪中钪含量的方法回收率,不符合国家标准gb/t27404-2008《实验室质量控制规范食品理化检测》中对回收率试验的规定。
[0102]
本发明进行了精密度以及加标回收率实验,检测结果稳定性、准确性符合相关要求。此外,本发明的检测方法工艺合理,安全可靠,操作简便,费用较低,满足生产需要。
[0103]
对比例2
[0104]
对比例2与实施例1的区别在于氢氧化钠溶液的浓度不同:实施例1的氢氧化钠为0.25:8-15,对比例2的氢氧化钠为0.25:7,对比例2过滤氢氧化钪沉淀使钪与氟分离后,钪含量测试精密度差,不符合gb/t27404-2008中的表f.2(实验室内变异系数)。对比例2将钪标加入样品中,检测加标回收率结果均低于65%,不符合国家标准gb/t27404-2008《实验室质量控制规范食品理化检测》中对回收率试验的规定。
[0105]
对比例3
[0106]
对比例3与实施例1的区别在于氢氧化钠溶液的浓度不同:实施例1的氢氧化钠为0.25:8-15,对比例3的氢氧化钠为0.25:16,对比例3在过滤氢氧化钪沉淀时滤纸被烫卷,无法过滤而无法实现钪离子与氟离子的分离,氟离子干扰edta-2na滴定钪的终点判定(滴定时终点无法达到亮黄色,见【图3】)。
[0107]
以上内容仅为本发明较佳的具体实施方式,不能认定本发明的具体实施方式只局限于这些说明,对于任何熟悉本技术领域的技术人员,在为未脱离本发明的技术方案下得出的其他实施方式,均应涵盖在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1