一种分离测定硫酸奈替米星注射液中有关物质的方法与流程
1.本发明属于药物分析技术领域,特别涉及一种分离测定硫酸奈替米星注射液中有关物质的方法。
背景技术:
2.奈替米星是一类半合成氨基糖苷类抗生素,从结构上分析是西索米星1-n-乙基衍生物,又称为乙基西索米星。奈替米星的抗菌谱广,对革兰阴性菌以及部分革兰阳性菌均有较强的抗菌活性。奈替米星对耐庆大霉素、西索米星、妥布霉素的菌群仍有抗菌活性,且其耳肾毒性低于以上氨基糖苷类抗生素,临床应用为其硫酸盐。硫酸奈替米星的化学结构式如下:
[0003][0004]
根据硫酸奈替米星的合成工艺及主要降解杂质情况,本品的起始原料及主要工艺杂质硫酸西索米星(杂质a)、主要降解杂质(杂质b)是影响硫酸奈替米星注射液纯度和质量的关键性杂质,杂质a和杂质b化学结构式如下:
[0005]
杂质a(硫酸西索米星)
[0006][0007]
杂质b(1-n-乙基加洛糖胺硫酸盐)
[0008][0009]
硫酸奈替米星分子结构中仅有一个双键,在低波长处有较弱的紫外吸收,其相关降解产物(杂质b)无紫外吸收,故美国药典中的高效液相色谱法不适用本品有关物质的检测。有文献报道中国药典中的蒸发光散射检测器法重现性较差,杂质灵敏度较低。欧洲药典采用离子色谱-脉冲安培检测器方法,奈替米星与辅料中的苯甲醇无法有效分离,且ep药典方法在试验进行过程中,流动相含有大量的盐,系统平衡非常困难。
[0010]
因此,亟待一种测定方法实现硫酸奈替米星及有关物质的测定分离,该方法在依折麦布的合成及其质量控制方面具有重要应用价值。
技术实现要素:
[0011]
为解决上述技术问题,本发明的目的在于提供一种分离测定硫酸奈替米星注射液中有关物质的方法。该方法专属性和针对性更强,对硫酸奈替米星注射液中存在的杂质可以进行准确的定量检测,并控制杂质限度,从而更有效地控制产品质量,为有关物质研究提供一定的基础。
[0012]
为实现上述技术目的,达到上述技术效果,本发明通过以下技术方案实现:
[0013]
一种分离测定硫酸奈替米星注射液中有关物质的方法,包括如下步骤:
[0014]
(1)配制供试品溶液
[0015]
取硫酸奈替米星注射液适量,加流动相a溶解并稀释,制成供试品溶液;
[0016]
(2)配制对照品溶液
[0017]
a.配制硫酸奈替米星对照品储备液:取硫酸奈替米星对照品适量,加稀释液溶解并稀释,制成硫酸奈替米星对照品储备液;
[0018]
b.配制杂质a对照品储备液:取杂质a对照品适量,加稀释液溶解并稀释,制成杂质a对照品储备液;
[0019]
c.配制杂质b对照品储备液:取杂质b对照品适量,加稀释液溶解并稀释,制成杂质b对照品储备液;
[0020]
d.配制对照品溶液:精密量取硫酸奈替米星对照品储备液、杂质a对照品储备液和杂质b对照品储备液各适量,置于同一容器中,加流动相a稀释,制成对照品溶液;
[0021]
(3)色谱分析测定
[0022]
精密量取供试品溶液和对照品溶液,分别注入离子色谱仪,记录液相色谱图,并根据液相色谱图分析测定硫酸奈替米星含量、杂质a含量、杂质b含量、未知单个杂质含量和总杂质含量。
[0023]
进一步的,所述流动相a为0.05%五氟丙酸溶液-乙腈,体积比为97:3。
[0024]
进一步的,步骤(1)中,供试品溶液浓度为0.5~1mg/ml。
[0025]
进一步的,步骤(2)中,硫酸奈替米星对照品储备液中的硫酸奈替米星对照品浓度为1~1.5mg/ml,杂质a对照品储备液中的杂质a对照品浓度为1.4~1.6mg/ml,杂质b对照品储备液中的杂质b对照品浓度为1.4~1.6mg/ml。
[0026]
进一步的,步骤(2)中,对照品溶液中的硫酸奈替米星对照品浓度为7~8μg/ml,对照品溶液中的杂质a对照品浓度为7~8μg/ml,对照品溶液中的杂质b对照品浓度为14~16μg/ml。
[0027]
进一步的,所述离子色谱仪采用的色谱柱为welch lp-c18柱,色谱柱利用十八烷基硅烷键合硅胶作为填充剂,色谱条件为:色谱柱中的流动a为0.05%五氟丙酸溶液-乙腈(97﹕3),流动相b为0.2mol/l三氟醋酸溶液-乙腈(97﹕3);流速为0.8ml/min,检测波长为232nm,色谱柱柱温为35℃,进样量为30μl。
[0028]
进一步的,步骤(3)中,采用梯度洗脱,用积分脉冲安培电化学检测器检测;检测电极为金电极;参比电极为ag/agcl复合电极,钛合金对电极;四波形检测电位。
[0029]
本发明的有益效果是:
[0030]
本发明设计了供试品溶液和对照品溶液,通过配合,根据液相色谱图分析测定硫酸奈替米星片单杂含量和总杂含量。
[0031]
本发明采用离子色谱法分离测定硫酸奈替米星片中有关物质,实现了硫酸奈替米星与2种杂质的分离,灵敏度高,可实现基线分离,专属性好,杂质含量测定准确,操作简捷,主成分与个杂质之间的分离度均大于10.0,分离度量好,可用于硫酸奈替米星注射液的质量控制,应用前景广阔。
附图说明
[0032]
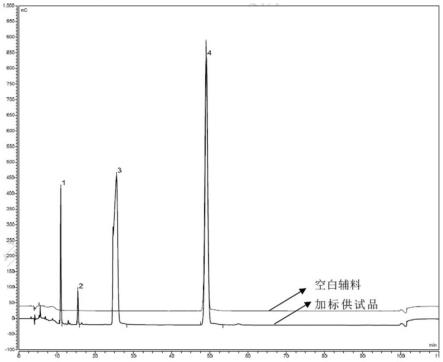
图1为本发明实施例中的对照品溶液的液相色谱图。
[0033]
图2为本发明实施例中的供试品溶液的液相色谱图。
具体实施方式
[0034]
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0035]
本发明提供了一种分离测定硫酸奈替米星注射液中有关物质的方法,包括如下步骤:
[0036]
(1)配制供试品溶液
[0037]
取硫酸奈替米星注射液适量,加流动相a溶解并稀释,制成供试品溶液;
[0038]
(2)配制对照品溶液
[0039]
a.配制硫酸奈替米星对照品储备液:取硫酸奈替米星对照品适量,加稀释液溶解并稀释,制成硫酸奈替米星对照品储备液;
[0040]
b.配制杂质a对照品储备液:取杂质a对照品适量,加稀释液溶解并稀释,制成杂质a对照品储备液;
[0041]
c.配制杂质b对照品储备液:取杂质b对照品适量,加稀释液溶解并稀释,制成杂质
b对照品储备液;
[0042]
d.配制对照品溶液:精密量取硫酸奈替米星对照品储备液、杂质a对照品储备液和杂质b对照品储备液各适量,置于同一容器中,加流动相a稀释,制成对照品溶液;
[0043]
(3)色谱分析测定
[0044]
精密量取供试品溶液和对照品溶液,分别注入离子色谱仪,记录液相色谱图,并根据液相色谱图分析测定硫酸奈替米星含量、杂质a含量、杂质b含量、未知单个杂质含量和总杂质含量。
[0045]
其中,所述流动相a为0.05%五氟丙酸溶液-乙腈,体积比为97:3。
[0046]
步骤(1)中,供试品溶液浓度为0.5~1mg/ml。
[0047]
步骤(2)中,硫酸奈替米星对照品储备液中的硫酸奈替米星对照品浓度为1~1.5mg/ml,杂质a对照品储备液中的杂质a对照品浓度为1.4~1.6mg/ml,杂质b对照品储备液中的杂质b对照品浓度为1.4~1.6mg/ml。
[0048]
步骤(2)中,对照品溶液中的硫酸奈替米星对照品浓度为7~8mg/ml,对照品溶液中的杂质a对照品浓度为7~8mg/ml,对照品溶液中的杂质b对照品浓度为14~16mg/ml。
[0049]
其中,所述离子色谱仪采用的色谱柱为welch lp-c18柱,色谱柱利用十八烷基硅烷键合硅胶作为填充剂,色谱条件为:色谱柱中的流动a为0.05%五氟丙酸溶液-乙腈(97﹕3),流动相b为0.2mol/l三氟醋酸溶液-乙腈(97﹕3);流速为0.8ml/min,检测波长为232nm,色谱柱柱温为35℃,进样量为30μl。
[0050]
步骤(3)中,采用梯度洗脱,用积分脉冲安培电化学检测器检测,检测电极为金电极,参比电极为ag/agcl复合电极,钛合金对电极,四波形检测电位。
[0051]
下面利用一个具体实施例说明本发明的方法。
[0052]
1、仪器与条件
[0053]
仪器:离子色谱仪
[0054]
色谱柱:十八烷基键合硅胶为填充剂(welch lp-c18 250mm
×
4.6mm 5μm或效能相当的色谱柱)
[0055]
流动相(v:v):以0.05%五氟丙酸溶液-乙腈(97﹕3)为流动相a,以0.2mol/l三氟醋酸溶液-乙腈(97﹕3)为流动相b,进行梯度洗脱。
[0056]
柱温:35℃
[0057]
流速:0.8ml/min
[0058]
检测器:积分脉冲安培电化学检测器
[0059]
检测电极:金电极(推荐使用3mm直径)
[0060]
参比电极:ag/agcl复合电极,钛合金对电极,四波形检测电位
[0061]
进样体积:30μl。
[0062]
2、实验步骤
[0063]
2.1、取硫酸奈替米星注射液适量,加流动相a溶解并稀释制成每1ml中约含硫酸奈替米星0.75mg的溶液;
[0064]
2.2、取硫酸奈替米星对照品、杂质a对照品及杂质b对照品适量,精密称定,加乙腈-水(1:1)溶解并稀释制成每1ml约含硫酸奈替米星1.25mg、杂质a1.5mg及杂质b1.5mg的溶液,分别作为硫酸奈替米星对照品储备液、杂质a对照品储备液及杂质b对照品储备液;精
密量取各各储备液适量,用流动相a稀释制成每1ml溶液约含硫酸奈替米星7.5μg、杂质a7.5μg及杂质b 15μg的溶液;
[0065]
2.3、用十八烷基键合硅胶为填充剂(welch lp-c18 250mm
×
4.6mm 5μm或效能相当的色谱柱);以0.05%五氟丙酸溶液-乙腈(97:3)为流动相a,以0.2mol/l三氟醋酸溶液(含0.1mol/l氢氧化钠溶液和0.05%w/v无水硫酸钠)-乙腈(96:4)为流动相b,按下表1进行梯度洗脱,流速为每分钟0.8ml;柱温为35℃;用积分脉冲安培电化学检测器检测;检测电极为金电极(推荐使用3mm直径);参比电极为ag/agcl复合电极,钛合金对电极;四波形检测电位(见下表2),柱后加碱(50%氢氧化钠溶液1
→
25,含0.005mol/l的乙二胺四乙酸二钠溶液,推荐流速每分钟0.3ml);进样体积30μl。
[0066]
表1
[0067][0068]
表2
[0069][0070][0071]
系统适用性要求:对照品溶液的液相色谱图中出峰顺序依次为杂质b(峰1)、杂质a(峰2)与硫酸奈替米星(峰3),各成分峰之间的分离度应不小于10.0。
[0072]
2.4、精密量取供试品溶液和对照品溶液,分别注入离子色谱仪,记录液相色谱图,结果如图1和图2,并根据液相色谱图按照外标法以峰面积测定有关物质含量。
[0073]
本发明未具体描述的部分采用现有技术即可,在此不做赘述。
[0074]
以上所述仅为本发明的实施例,并非因此限制本发明的专利范围,凡是利用本发明说明书内容所作的修改或等效变换,或直接或间接运用在其他相关的技术领域,均同理包括在本发明的专利保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1