基于一步式固相萃取快速测定动物性食品中氨丙啉残留量的方法与流程
1.本发明属于食品分析技术领域,具体涉及一种基于一步式固相萃取快速测定动物性食品中氨丙啉残留量的方法。
背景技术:
2.氨丙啉又名安保宁,属于抗硫胺素类抗球虫药物,具有高效广谱的杀菌能力,在鸡、牛、羊、兔等多种动物的养殖过程中被广泛使用。由于球虫病具有传染性,可明显降低动物的生产效能,甚至引发动物大批死亡,给养殖者带来重大经济损失。为达到抗球虫的目的,养殖过程中存在不科学、不规范用药,以及不严格执行休药期等现象。上述因素均可导致动物性食品中氨丙啉残留,进而影响人体对硫胺素的吸收,严重时可能造成韦尼克脑病。因此日本、加拿大、韩国、美国等许多国家都对动物性食品中氨丙啉最大残留限量做了明确规定,我国gb 31650-2019中也明确规定了牛、鸡、火鸡的肌肉、肝、肾、脂肪以及鸡蛋中氨丙啉的最大残留量。因此,建立动物性食品中氨丙啉残留量的分析方法,全面评估食品中氨丙啉的残留现状,对保障消费者饮食安全意义重大。
3.已报道的氨丙啉分析方法主要包括毛细管电泳法、荧光光谱法、分光光度法、比色法、液相色谱法和液质联用法等。液质联用法将液相色谱快速、高效的分离能力和质谱检测器的定性、定量功能力相结合,具有灵敏度高、抗干扰能力强等特点,已成为当前兽药残留的主要检测手段。关于氨丙啉的检测,我国现行的检测标准主要包括《牛可食性组织中氨丙啉残留量的测定》(gb 31613.1-2021)、《出口动物源食品中抗球虫药物残留量检测方法 液相色谱-质谱/质谱法》(sn/t 3144-2011)和《出口动物源食品中氨丙啉残留量的测定液相色谱-质谱/质谱法》(sn/t 4583-2016)。三项标准的前处理过程包括目标物提取、溶剂转换、固相萃取净化、浓缩等多个步骤,过程复杂耗时,不适于监管的快速检测要求,且gb 31613.1基质涵盖不全,无法满足gb 31650的监测需求,因此亟需建立食品中氨丙啉的快速高效检测方法以满足监管需求。
技术实现要素:
4.本文首次引入一步式固相萃取柱净化,结合亲水作用色谱柱进行分离,串联质谱检测,建立了动物性食品中氨丙啉残留的快速检测方法。对提取溶剂进行优化,酸化乙腈溶液可实现目标物离子化,减弱了目标物与固相萃取柱之间的相互作用,保证了目标物的回收率;对净化过程进行优化,采用的prime hlb固相萃取柱可去除蛋白、磷脂等动物性食品中典型基质干扰物,且净化过程无需填料的活化与平衡,提取溶液直接上样即可获得洁净的样品溶液,避免了常规spe的淋洗、洗脱等步骤,大大节省了样品前处理时间。同时样品净化液中由于含有较高比例有机相,可直接应用于亲水作用色谱的进样分析,进而再次省略了溶剂转化步骤,较现有标准方法,实验效率显著提高。结合方法学验证结果,本方法具有灵敏度高、重现性好、操作便捷等特点,可以为动物性食品中氨丙啉的快速检测提供技术支
持。
5.为实现上述目的,本发明采用下述技术方案:一种基于一步式固相萃取快速测定动物性食品中氨丙啉残留量的方法,包括以下步骤:(1)样品提取:称取样品,加入提取剂,涡旋混匀,超声离心;加入提取剂重复提取,合并2次提取溶液,定容,摇匀备用;(2)样品净化:提取液通过prime hlb固相萃取柱,收集流出液,涡旋混匀后微孔滤膜过滤,供仪器测定;(3)待测样品净化液进行超高液相色谱-串联质谱检测。
6.进一步地,提取剂为含0.2%甲酸的80%乙腈溶液。
7.进一步地,样品提取的具体过程为:称取2.5 g样品于50 ml离心管中,加入20 ml含0.2%甲酸的80%乙腈水溶液,涡旋混匀1min,超声10 min,8500 r/min进行离心2 min,上层清液转移至50 ml容量瓶中;再加入20 ml提取剂重复提取1次,合并2次提取溶液,用提取剂定容至刻度。
8.进一步地,样品净化的具体过程为:准确移取3.0 ml上述提取液,注入prime hlb固相萃取柱中,使溶液全部通过固相萃取柱,收集流出液;抽干固相萃取柱,涡旋混匀后,经0.22 μm微孔滤膜过滤,供仪器测定。
9.进一步地,超高液相色谱的条件为:色谱柱: waters acquity uplc beh hilic,尺寸为100
×
2.1mm,1.7μm;流动相:a为含0.05%甲酸的5 mmol/l甲酸铵溶液,b为乙腈;流速:0.30 ml/min;柱温:40 ℃;进样体积:2 μl;洗脱梯度为:。
10.进一步地,质谱条件为:电喷雾离子源,正离子扫描;检测方式:多反应监测;电喷雾电压:5500 v;雾化器压力:55 l/h;气帘气压力:45 l/h;辅助气压力:50 l/h;碰撞气压力:7 l/h;离子源温度:550 ℃;辅助气和雾化气为压缩空气,碰撞气和气帘气为高纯氮气;定性离子对、定量离子对、采集时间、去簇电压及碰撞能量为:。
11.本发明的有益效果为:本发明基于一步式固相萃取,形成了动物性食品中氨丙啉的快速测定方法。方法采用80%乙腈溶液(含0.2%甲酸)作为提取剂,高比例乙腈可使蛋白变性,滤液澄清,为spe的净化速度提供保证;同时0.2%甲酸的存在也显著提高了氨丙啉的回收率。提取液一步完成
净化,无需spe前的溶剂转换,也省去了活化、淋洗、洗脱、洗脱液的浓缩以及复溶等5个步骤,与现有检测标准相比,前处理效率提升3-5倍。本方法以beh hilic色谱柱进行分离,保留时间适中,色谱峰对称,响应高,灵敏度比现有检测标准提高2-5倍;方法的回收率和重现性俱佳,适用于批量样本中氨丙啉的快速检测。
附图说明
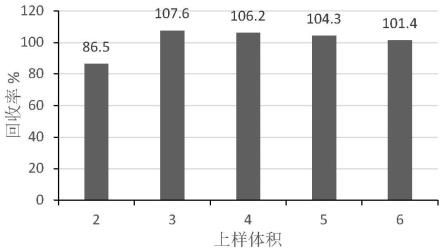
12.图1为不同净化柱对净化效果比较图;图2 为不同上样体积的回收率;图3 为不同上样体积的基质效应;图4为2.5 ng/ml牛肾标准溶液(a)、牛肾空白样品(b)、牛肾定量限加标样品(c)的特征离子色谱图。
具体实施方式
13.为了使技术领域的人员更好地理解本发明方案,并使本发明的上述目的、特征和优点更加明显易懂,下面结合实施例对本发明作进一步的详细说明。
14.实施例11 实验部分1.1 仪器、试剂与材料ab sciex triple quad 6500+超高效液相色谱-串联质谱仪(配有电喷雾电离源)(美国ab sciex公司);3-18ks高速冷冻离心机(德国sigma公司);secura1102-1cn-sqp电子天平(赛多利斯科学仪器有限公司);ms3型旋涡混合器(德国ika公司);dtc-33超声清洗机(湖北鼎泰恒胜科技设备有限公司);milli-q超纯水系统(美国millipore公司)。
15.乙腈(色谱纯,霍尼韦尔贸易(上海)有限公司),甲酸铵、甲酸(色谱纯,美国sigma-aldrich公司);盐酸氨丙啉标准品(cas号:137-88-2,纯度为98.6%)购于北京曼哈格生物科技有限公司;oasis prime hlb固相萃取柱(200 mg/6ml,美国waters公司);beh hilic 色谱柱(100
×
2.1mm,1.7 μm,美国waters公司)。
16.样品处理1.2.1提取称取2.5 g样品(精确至0.01g)于50 ml离心管中,加入20 ml提取剂(80%乙腈溶液(含0.2%甲酸)),涡旋混匀1min,超声10 min,8500 r/min进行离心2 min,上层清液转移至50 ml容量瓶中。再加入20 ml提取剂重复提取1次,合并2次提取溶液,用提取剂定容至刻度,摇匀备用。
17.1.2.2净化准确移取3.0 ml上述提取液,注入prime hlb固相萃取柱,使溶液全部通过固相萃取柱,收集流出液。抽干固相萃取柱,涡旋混匀后,经0.22 μm微孔滤膜过滤,供仪器测定。
18.仪器分析条件1.3.1 色谱条件a)色谱柱: waters acquity uplc beh hilic (100
×
2.1mm,1.7μm);b)流动相:a为含0.05%甲酸的5 mmol/l甲酸铵溶液,b为乙腈,洗脱梯度见表1;
c)流速:0.30 ml/min;d)柱温:40 ℃;e)进样体积:2 μl。
19.表1 uplc梯度洗脱程序1.3.2 质谱条件电喷雾离子源(esi源),正离子扫描;检测方式:多反应监测(mrm);电喷雾电压:5500 v;雾化器压力:55 l/h;气帘气压力:45 l/h;辅助气压力:50 l/h;碰撞气压力:7 l/h;离子源温度:550 ℃。辅助气和雾化气为压缩空气,碰撞气和气帘气为高纯氮气。定性离子对、定量离子对、采集时间、去簇电压及碰撞能量见表2。
20.表2 氨丙啉的质谱参数* quantitative ion2 结果与讨论2.1 前处理方法的优化2.1.1 提取溶剂的选择氨丙啉易溶于水、乙醇和甲醇,不溶于氯仿。磷酸盐、甲醇、乙腈和三氯乙酸-乙腈溶液是氨丙啉测定常用的提取溶剂。本方法以牛肾为验证样品,考察了磷酸盐、甲醇、乙腈、80%乙腈水(含0.2%甲酸)和1%三氯乙酸-乙腈(3:7)五种提取溶剂对氨丙啉的提取效果。实验表明,磷酸盐提取后溶液浑浊,其余4种提取溶液澄清,且回收率均在85%以上。本方法拟采用固相柱的通过模式进行净化,提取溶剂通过spe后直接过膜上机测定,无溶剂转化过程。纯甲醇和纯乙腈对磷脂等弱极性杂质的洗脱能力太强,影响净化效果;1%三氯乙酸-乙腈(3:7)中三氯乙酸含量较高,通过spe后直接进质谱,有影响后续的负离子化合物分析的风险。因此,方法优先选择80%乙腈溶液(含0.2%甲酸)作为提取溶剂。
21.本研究又对提取溶剂中甲酸的浓度进行了优化,验证了80%乙腈水溶液中甲酸分别占0%、0.1%、0.2%、0.5%和2%时氨丙啉的回收率情况。结果显示,不加甲酸时,氨丙啉回收率为66.1%;当甲酸浓度为0.1%时,回收率为82.3%;当甲酸浓度≥0.2%时,回收率均高于90%并趋于稳定。原因可能是当提取溶剂中甲酸量不足时,氨丙啉部分呈分子状态,与spe之间存在反相吸附作用,从而保留在spe上,造成回收率偏低;当提取溶剂中甲酸浓度≥0.2%之后,氨丙啉全部呈阳离子状态,与spe之间作用力较弱,无法保留在spe上,从而提高了方法
的回收率。综上,将80%乙腈溶液(含0.2%甲酸)确定为本方法的提取溶剂。
22.2.1.2 固相萃取柱的选择为降低动物性食品中基质影响,减少对质谱和色谱柱的污染,本研究考察了hlb、prime hlb和emr-lipid三种spe对氨丙啉的净化情况。其中hlb采用为目标物保留模式,prime hlb和emr-lipid采用目标物通过模式。实验条件如下:(1)hlb固相萃取(60 mg,3 ml):按照1.2.1步骤进行提取,准确移取3 ml样品提取溶液,40℃氮吹至近干,用5 ml磷酸盐溶液(ph=6.0)复溶,复溶液全部通过hlb固相萃取柱(提前用3 ml甲醇、3 mlh2o活化),再用3 ml h2o淋洗,最后加入甲醇2 ml进行洗脱。洗脱液于40℃氮吹干后,准确加入1 ml初始流动相进行复溶,涡旋混匀3 min,过0.22 μm滤膜过滤,供仪器分析测定。
23.(2)通过式固相萃取:按照1.2.1步骤进行提取,分别取3 ml样品提取溶液,注入prime hlb(200 mg,6 ml)和 emr-lipid(300 mg,3ml)中,收集样品净化液于15 ml离心管中,涡旋混匀后,过0.22 μm滤膜过滤,供仪器测定。
24.三种净化柱对回收率的影响详见图1。由图1可知,hlb和prime hlb回收率均在90%以上,emr-lipid对氨丙啉有部分吸附,回收率为74.4%。prime hlb是在hlb的基础上进行了创新,与hlb相比,保持了良好的水可浸润性,同时去除蛋白和磷脂的能力更强;特殊的筛板设计、填料和制作工艺,可使流速更快更稳定。本试验选择prime hlb进行一步式净化,该净化方式无需溶剂转换,省去了spe的活化、淋洗、洗脱以及洗脱液的氮吹浓缩和复溶,步骤简单、检验效率更高。
25.本研究进一步对60 mg、200 mg和500 mg三种规格型号的prime hlb固相萃取柱对回收率和基质效应的影响进行了考察。60 mg的spe回收率97.8%,填料少净化效果差,基质效应为59.9%;200 mg的spe回收率107.6%,基质效应为30.1%;填料为500 mg的spe上目标物部分保留,回收率仅为78.2%。综合考虑,本方法最终选择prime hlb(200 mg,6 ml)的固相萃取柱进行氨丙啉的净化。
26.2.1.3 上样体积的优化对于通过式固相萃取,上样体积会同时影响回收率和净化效果,所以本方法比较了上样体积对回收率和基质效应两方面的影响,结果如图2和图3所示。图2表明,上样体积为2 ml时回收率稍低,为86.5%,上样体积≥3 ml时回收率稳定,均在100%左右;图3表明,上样体积越大,净化效果越差,基质效应增强越显著;最终上样体积确定为3 ml。
27.仪器分析条件的优化2.2.1 质谱条件的优化氨丙啉结构中含有季铵基团,极性很强,适合电喷雾离子源。在酸性条件下,氨丙啉分子结构中的氨基易结合质子形成正离子,因此选择正离子电离模式。将50 ng/ml的氨丙啉标准溶液装入注射泵中,esi源正离子模式下,进行全扫描,获得氨丙啉的分子离子峰[m-cl+h]
+
为242.9;以m/z 242.9为母离子,寻找信号强而稳定的二级碎片离子,得到m/z 150.0和m/z 94.0两个碎片离子;在mrm模式下,继续优化去簇电压、碰撞能等质谱参数,最优质谱条件见表1。
[0028]
2.2.2 色谱条件的优化2.2.2.1 色谱柱的选择
氨丙啉属于季胺类化合物,在传统的反相色谱柱保留弱,本研究考察了hss t3(100 mm
×
2.1 mm,1.8 μm)、beh amide(100 mm
×
2.1 mm,1.7 μm)和beh hilic (100 mm
×
2.1 mm,1.7 μm)三种类型的色谱柱。hss t3与普通c
18
柱相比,能够为极性化合物提供更强的保留。实验表明,乙腈比例低至2%的情况下氨丙啉的保留时间为1.21min,此时待测样本中强极性化合物出峰比较集中,易干扰目标物的定性定量。剩余2款亲水型色谱柱都能为氨丙啉提供适合的色谱保留,beh hilic色谱柱上的峰形更对称,且响应值比beh amide高2倍。beh hilic柱使用未键合的亚乙基桥杂化颗粒(beh)颗粒,该填料在ph为1-9范围内,化学性质稳定,与氨丙啉之间存在氢健作用和偶极间作用力,非常适合氨丙啉的测定。zic-hilic 色谱柱是在硅胶表面键合了一个磺基甜菜碱结构的两性离子键合相,适宜的ph范围为3-8。采用 sequant zic-hilic ( 150 mm
ꢀ×ꢀ
2.1mm,5 μm) 色谱柱分析氨丙啉时,氨丙啉与固定相之间除氢健作用和偶极间作用力外,还存在氨丙啉与zic-hilic固定相上电荷之间的静电作用。zic-hilic与分析物之间作用机理复杂,适用ph范围窄,在方法重现性方面挑战更大,因此,本方法选择beh hilic色谱柱进行氨丙啉的分析。
[0029]
2.2.2.2 流动相的选择氨丙啉在正离子模式下易电离,酸性流动相能够提高氨丙啉的质谱响应。本研究比较了0.05%甲酸溶液-乙腈、5 mmol/l甲酸铵溶液-乙腈、含0.05%甲酸的5 mmol/l甲酸铵溶液-乙腈、含0.05%甲酸的10 mmol/l甲酸铵溶液-乙腈、含0.1%甲酸的5 mmol/l甲酸铵溶液-乙腈和含0.2%甲酸的5 mmol/l甲酸铵溶液-乙腈等6种流动相组合。实验表明,乙腈-5 mmol/l甲酸铵(含0.05%甲酸)为流动相时,氨丙啉峰形和响应值最佳,保留时间为3.22 min。继续增加甲酸铵的浓度至10 mmol/l时,氨丙啉的响应值反而降低了8倍;乙腈-5 mmol/l甲酸铵体系中继续增加甲酸的浓度,目标物的保留时间和响应值均无明显变化,综合考虑选择乙腈-5 mmol/l甲酸铵(含0.05%甲酸)为流动相。
[0030]
方法学考察2.3.1 线性范围、检出限和定量限将基质匹配标准工作溶液(0.25 ng/ml、0.5 ng/ml、2.5 ng/ml、10.0 ng/ml、25.0 ng/ml、50.0 ng/ml、100 ng/ml)进行uplc-ms/ms分析,获得标准溶液的质量色谱图。以氨丙啉峰面积(y)为纵坐标,以标曲浓度(x)为横坐标,绘制标准曲线,得到线性回归方程。10种食品基质的线性回归方程和相关系数详见表3。结果显示氨丙啉在0.25~100 ng/ml范围内呈良好的线性,相关系数(r2)均优于0.99。
[0031]
表3 10种食品基质中氨丙啉的线性范围、回归方程和相关系数
ꢀ
分别向上述10种空白样品中添加系列浓度的标准物质,以3倍信噪比,确定方法的检出限(lod),以10倍信噪比,确定方法的定量限(loq)。氨丙啉的检出限和定量限分别2.0 μg/kg和5.0 μg/kg,各基质灵敏度均可满足监管需求。牛肾基质匹配标准溶液(2.5 ng/ml)、牛肾空白样品和牛肾定量限水平样品特征离子色谱图如图4所示。
[0032]
2.3.2回收率与精密度选择鸡肉、鸡肝、牛奶等10种空白基质,进行低中高水平(loq、10loq和100loq)的加标回收试验,每个浓度水平平行测定6次,评价方法回收率和精密度,结果详见表4。结果表明,氨丙啉在10种基质中平均回收率为86.1~108.5%,rsd为2.2~8.4%。
[0033]
表4 不同基质中氨丙啉的回收率与精密度(n=6)
ꢀ
2.4 实际样品测定应用本文所建立的方法对市售鸡肉10批次、鸡肝5批次、鸡肾5批次、鸡胗3批次、鸡
蛋10批次、牛肉5批次、牛肝5批次、牛肾3批次、牛奶10批次以及牛脂肪1批次,共计56批次样品进行了氨丙啉的检测,有2批次鸡蛋样品中检出氨丙啉,检出值分别为 56.6 μg/kg和20.8 μg/kg,其他样品均未检出。
[0034]
方法准确性考察用本方法和sn/t 4583-2016对2.4中有氨丙啉检出的2批次鸡蛋同时进行检测,以验证本方法的准确性,检测结果见表5。结果显示2个鸡蛋样品中的氨丙啉含量和标准方法测定结果基本一致,相对偏差分别为6.19%和9.57%,可见该方法准确性较好。由于本方法操作步骤简单,测定结果的平行性更好。
[0035]
表5 本方法与标准方法的比对结果3 结论本文基于一步式固相萃取,形成了动物性食品中氨丙啉的快速测定方法。方法采用80%乙腈溶液(含0.2%甲酸)作为提取剂,高比例乙腈可使蛋白变性,滤液澄清,为spe的净化速度提供保证;同时0.2%甲酸的存在显著提高了氨丙啉的回收率。提取液一步完成净化,spe净化前无需溶剂转换,也省去了活化、淋洗、洗脱、洗脱液的浓缩以及复溶等5个步骤,与现有检测标准相比,前处理效率提升3-5倍。本方法以beh hilic色谱柱进行分离,保留时间适中,色谱峰对称,响应高,灵敏度比现有检测标准提高2-5倍;方法的回收率和重现性俱佳,适用于批量样本中氨丙啉的快速检测。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1