中药材中可挥发弱极性-非极性有机污染物的前处理试剂及其方法和检测应用与流程
本发明涉及一种有机污染物的前处理试剂,尤其是涉及一种中药材中可挥发弱极性-非极性有机污染物的前处理试剂及其方法和检测应用。
背景技术:
1、弱极性-非极性有机污染物如pcbs、多环芳烃、有机氯类及其它弱极性农药等,除了少部分在apci有信号外,绝大多数没有电喷雾esi信号,无法依靠高灵敏的lc-ms/ms分析。它们具有可气化性和热稳定性共性,可通过气相色谱-质谱gc-ms或气相色谱-质谱联用gc-ms/ms检测方法进行分析。相对单一gc-ecd方法,可大幅缩短分析时间,而且同时具有筛选和确证的功能,尤其gc-ms/ms进一步提升了特异性和灵敏度等性能,成为了此类残留检测的最受欢迎的分析手段,在果蔬检测上得到了广泛使用。目前,弱极性-非极性有机污染物的前处理法通常以盐析乙腈提取和固相分散萃取净化(d-spe)为核心的quechers方法为主。另外,gcb/nh2复合spe小柱和gpc(凝胶渗透色谱)也是常有的净化手段。然而,对于基质复杂程度更高的中药材,尤其富含脂溶性的连翘、枸杞等,上述常规的前处理净化手段显得力不从心,不仅容易污染进样口、毛细色谱柱,而且离子源极易受污染带来灵敏度直线下降和数据的可靠性变差,甚至威胁到四级杆。仪器操作人员不得不花费大量精力进行清洗离子源和更换衬管、色谱柱。因此,如何克服复杂基质的干扰成为了中药材检测的迫切需要解决的难题。
技术实现思路
1、本发明所要解决的技术问题是提供一种通用性良好,净化效果强、准确率和回收率高的中药材中可挥发弱极性-非极性有机污染物的前处理试剂及其方法和检测应用。
2、本发明解决上述技术问题所采用的技术方案为:
3、1、一种中药材中可挥发弱极性-非极性有机污染物的前处理试剂,包括混合净化试剂和分散固相萃取填料,所述的混合净化试剂由以下成分及其质量百分比组成:33%的甲醇、65%的浓度为2mol/l且ph=4.8的乙酸铵溶液和2%的二甲基亚砜dmso组成;所述的分散固相萃取填料由无水硫酸镁、c18填料、c8填料、n-丙基乙二胺填料psa和中性氧化铝组成。混合净化试剂不仅可以浸泡中药材而且还能充分溶解强极性-中等极性组分,弱极性-非极性组分通过正己烷提取出来。无水硫酸镁用于吸水,其他粉末用于脱脂和去色素。
4、进一步,所述的c18填料的粒径为40μm,所述的c8填料的粒径为40μm,所述的n-丙基乙二胺填料的粒径为40μm,所述的中性氧化铝为100-200目。
5、进一步,所述的分散固相萃取填料中无水硫酸镁、c18填料、c8填料、n-丙基乙二胺填料psa和中性氧化铝的混合重量依次为200、200、200、200和400mg。
6、2、一种利用上述前处理试剂的中药材中可挥发弱极性-非极性有机污染物的前处理方法,包括以下步骤:
7、(1)第一步沉淀提取净化
8、称取待测样品于塑料离心管中,加入待测样品5倍体积的混合净化试剂,以10000rmp高速匀浆2min后,加入10ml正己烷,水平振荡30min,4500转/min离心5min,分成上下层,取下层清液于40℃下进行氮吹近干,再用10ml正己烷重复提取一次,合并两次提取的上层清液,将上层清液氮吹至近干;
9、(2)第二步固相分散萃取
10、在步骤(1)吹干后的试管中加入4ml乙腈,再倒入分散固相萃取填料,用不锈钢的刮刀(宽度5mm,长度200mm),充分刮磨和搅拌,使得试管底部和试管壁上所有提取物充分分散在填料中,加入4-5粒陶瓷均质子(3×6mm),漩涡混合5min后,放置于-40℃超低温冰箱中2h以上,取出后立即在0℃下,4000rmp离心2min,迅速通过0.2μm针式特富龙膜过滤到进样瓶中,进行gc-ms/ms分析。
11、3、一种中药材中可挥发弱极性-非极性有机污染物的gc-ms/ms检测方法,包括以下步骤:
12、(1)样品前处理
13、称取待测样品于塑料离心管中,加入待测样品5倍体积的混合净化试剂,以10000rmp高速匀浆2min后,加入10ml正己烷,水平振荡30min,4500转/min离心5min,分成上下层,取下层清液于40℃下进行氮吹近干,再用10ml正己烷重复提取一次,合并两次提取的上层清液,将上层清液氮吹至近干后,加入4ml乙腈,再倒入分散固相萃取填料,用不锈钢的刮刀,充分刮磨和搅拌,使得试管底部和试管壁上所有提取物充分分散在填料中,加入4-5粒陶瓷均质子,漩涡混合5min后,放置于-40℃超低温冰箱中2h以上,取出后立即在0℃下,4000rmp离心2min,迅速通过0.2μm针式特富龙膜过滤到进样瓶中,进行gc-ms/ms分析;
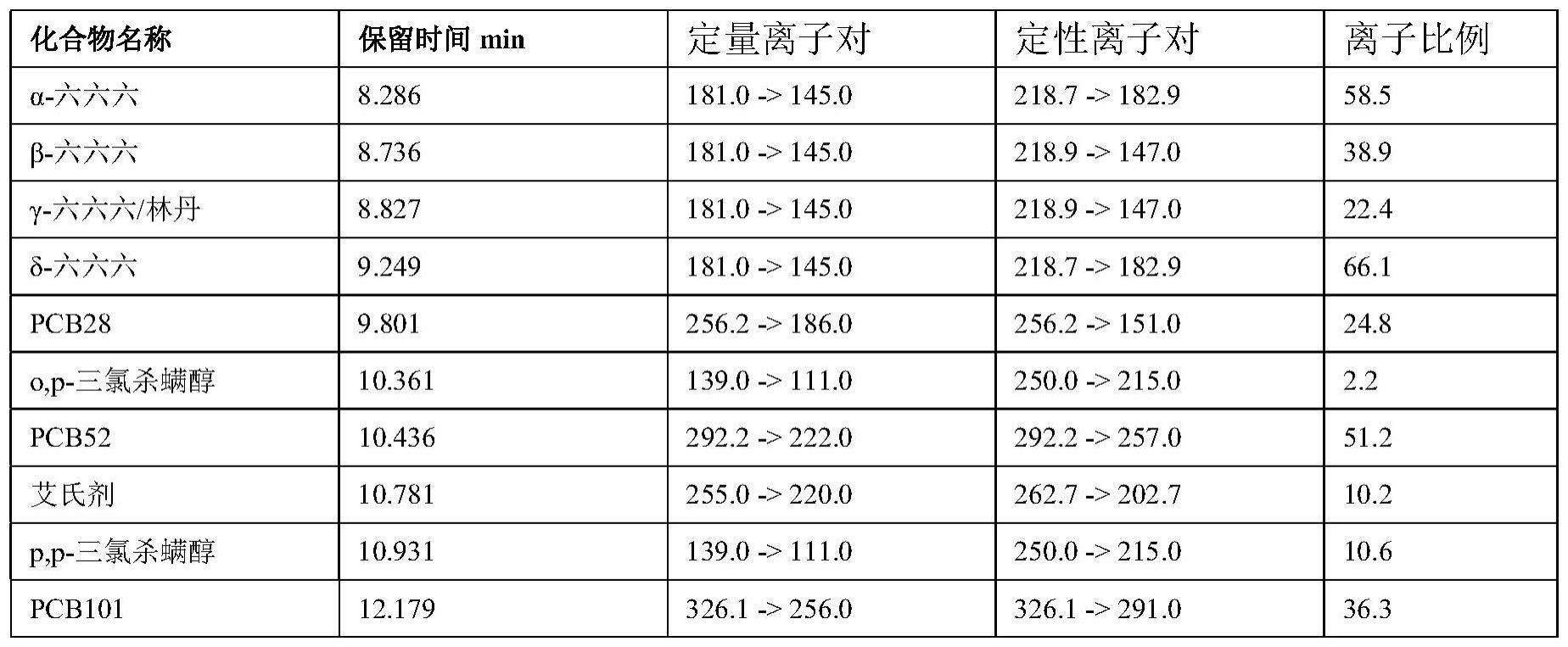
14、(2)基质标准曲线
15、在空白样品中依次添加0、20、40、200、400、1000μl标准工作溶液,静置15min,按步骤(1)前处理进行提取和净化,以浓度为横坐标,目标物的响应值为纵坐标,做基质加标标准曲线,作为试样处理液中待测物浓度定量的依据;所述的标准工作溶液中23种标准物质见下表1所示;
16、(3)气相色谱-三重四极杆串联质谱联用测定
17、色谱柱:含5%苯基亚芳基聚合物或5%苯基-甲基聚硅氧烷的弱极性毛细管;载气:氦气,流速为1ml/min;进样口温度:250℃;进样量:1μl,不分流进样,不分流时间为0.5min,溶剂延迟时间为5min;程序升温:70℃保持1min,以25℃/min升至180℃,再以8℃/min升至300℃,并保持8min;质谱参考条件:电离源:电子轰击源;电离能量:70ev;离子源温度:250℃;传输线温度:280℃;扫描方式:多通道选择离子监测模式;
18、表1 23种化合物的保留时间、定量和定性离子对见下表和离子比例
19、
20、
21、(3)浓度计算方法
22、样品中待测物的含量根据如下计算公式(1)得到:
23、
24、式中:x-试样中待测物含量,单位为μg/kg;
25、c-试样处理液中待测物浓度,根据基质标准曲线计算得到,单位为ng/ml;
26、m-试样质量,单位为g;
27、f—稀释因子。
28、进一步,所述的混合净化试剂由以下成分及其质量百分比组成:33%的甲醇、65%的浓度为2mol/l且ph=4.8的乙酸铵溶液和2%的二甲基亚砜dmso组成;所述的分散固相萃取填料由无水硫酸镁、c18填料、c8填料、n-丙基乙二胺填料psa和中性氧化铝组成。混合净化试剂不仅可以浸泡中药材而且还能充分溶解强极性-中等极性组分,弱极性-非极性组分通过正己烷提取出来。无水硫酸镁用于吸水,其他粉末用于脱脂和去色素。
29、进一步,所述的c18填料的粒径为40μm,所述的c8填料的粒径为40μm,所述的n-丙基乙二胺填料的粒径为40μm,所述的中性氧化铝为100-200目。
30、进一步,所述的分散固相萃取填料中无水硫酸镁、c18填料、c8填料、n-丙基乙二胺填料psa和中性氧化铝的混合重量依次为200、200、200、200和400mg。
31、与现有技术相比,本发明的优点在于
32、1、选择性强:样品通过混合净化溶剂和正己烷双相提取净化,强极性-中等极性组分的干扰物排除在提取液之外。而常规乙腈提取会带来大量极性-中极性的干扰物,不仅容易污染仪器,而且影响连续多次进样情况下的数据的可靠性。其次,其ph为4.8,既发挥了一定的沉淀蛋白的作用,且绝大多数目标物可获得更好的稳定性。
33、2、通用性强:适合弱极性-非极性范围的有机物污染物:如多氯联苯、六六六、滴滴涕、艾氏剂、三氯杀螨醇等,具体见表1所示。
34、3、基质分散固相萃取和超低温冷冻过滤法。针对复杂中药材,该混合填料具有良好的净化效果,与传统的固相萃取净化法相比,消耗的有机溶剂仅为其10%,大大节约时间和节约有机溶剂消耗,而且通用性和重复性更好。正己烷相对乙腈容易浓缩,有利于提高检测灵敏度。
35、综上所述,本发明中药材中可挥发弱极性-非极性有机污染物的前处理试剂及其方法和检测应用,采用极性分组策略,提升提取的选择性而降低后续净化难度,并结合基质分散固相萃取和超低温冷冻过滤等一系列简便通用的净化手段,成功开发植物性中药材里弱极性-非极性可挥发有机污染物的gc-ms/ms分析方法,取得了满意的效果。
- 还没有人留言评论。精彩留言会获得点赞!