基于UPLC-Q-TOF-MS/MS技术分析甘草多糖的方法与流程
本发明涉及检测,尤其涉及一种基于uplc-q-tof-ms/ms技术分析甘草多糖的方法。
背景技术:
1、基于uplc-q-tof-ms/ms技术分析甘草多糖的方法,目前已经有一些现有的技术和方法。以下是其中几种常用的方法:离子对色谱法,该方法通过添加离子对试剂,如三氟乙酸、甲酸等,以增强多糖化合物在uplc柱上的保留和分离效果。离子对试剂与多糖之间形成离子对复合物,提高了多糖的检测灵敏度和分离度。聚合物逆向相色谱法,该方法使用聚合物修饰的反相色谱柱,如聚合物甲基丙烯酸酯柱、聚合物乙烯基苯柱等,增强了多糖的保留和分离效果,并提高了质谱的信号强度。阴离子交换色谱法,该方法使用具有阴离子交换功能的色谱柱,如强阴离子交换柱,通过控制流动相中的ph值和离子强度,实现多糖的分离和定量分析。聚合物凝胶色谱法,该方法使用聚合物凝胶柱,如聚丙烯酰胺凝胶,通过多糖在凝胶柱中的分子大小和形状的差异,实现多糖的分离和纯化。质谱联用技术,将uplc与高分辨质谱仪联用,实现多糖的分子式、分子量和结构鉴定。通过质谱的高灵敏度和高分辨率,可以准确分析复杂的多糖混合物。
2、这些方法需要根据实际需要进行选择和优化,以获得高效、灵敏和准确的甘草多糖分析结果。
3、中国发明授权专利cn109553654b公开了一种从甘草中提取甘草甜素、甘草黄酮及甘草多糖的方法,所述从甘草中提取甘草甜素的方法,包括以下步骤:(1)将干燥的甘草根或根茎粉碎,投入渗漉器中,压紧,加水,在室温下,渗漉提取,得渗漉液;(2)超滤,收集超滤膜透过液;(3)上阴离子交换树脂柱,用洗脱剂洗脱,洗脱液减压浓缩,调节ph值至酸性,在室温下,搅拌析晶,离心过滤,冰水洗晶,干燥,得甘草甜素。该发明方法还公开了提取甘草黄酮及甘草多糖的方法。该发明方法所得甘草甜素、甘草黄酮和甘草多糖的纯度和收率高;该发明方法可提取多种活性成分,工艺过程可操作性强,成本低,不使用有毒有害化工溶剂、绿色环保,甘草资源高效综合利用,适宜于工业化生产。但是该发明提取方法分离性能差,不能完全提取甘草中的各种成分。
技术实现思路
1、有鉴于现有技术中甘草多糖的提取方法分离性能差的缺点,本发明所要解决的技术问题是提供一种分离性能好的基于uplc-q-tof-ms/ms技术分析甘草多糖的方法。
2、为了实现上述发明目的,本发明采用了如下的技术方案:
3、一种基于uplc-q-tof-ms/ms技术分析甘草多糖的方法:
4、步骤1、供试品溶液配制:称取甘草多糖,加入超纯水,超声处理溶解后,摇匀,制得甘草多糖溶液,经微孔滤膜过滤,得到供试品溶液;
5、步骤2、色谱条件:采用waters acquity uplc beh c18色谱柱(2.1mm×100 mm,1.7μm);采用流动相梯度洗脱,固定相为改性固定相;
6、步骤3、质谱条件:采用电喷雾离子源(esi),正、负离子2种模式扫描,质谱扫描;正离子模式下,负离子模式下,毛细管电压-4000~-5000kv,裂解电压-70~-90v,碰撞能量30~40ev;
7、步骤4、数据分析、采用analyst tf 1.8.1数据采集软件(美国 ab sciexi 公司),采集时间20~40min;采用sciex os 2.0.0软件进行数据处理(美国ab sciexi 公司),化学成分依据相对分子质量、离子碎片信息、保留时间,结合数据库及文献报道数据结果进行分析。
8、优选的,所述步骤1中为称取0.5~2重量份甘草多糖,加入超纯水,超声溶解后,摇匀,制得浓度为4~6mg/ml的甘草多糖溶液。
9、优选的,所述步骤1中微孔滤膜的孔径为0.2~0.25μm。
10、优选的,所述步骤2中流动相为乙腈和0.05~0.2wt%甲酸水溶液配置而成。
11、优选的,所述步骤2中流动相梯度洗脱参数为:0~2min,流动相由5%乙腈、0.05~0.2wt%甲酸水溶液补足100%组成;2~42min,流动相由5%~95%乙腈、0.05~0.2wt%甲酸水溶液补足100%组成;42~47min,流动相由95%乙腈、0.05~0.2wt%甲酸水溶液补足100%组成;47~47.1min,流动相由5%~95%乙腈、0.05~0.2wt%甲酸水溶液补足100%组成;47.1~50min,流动相由5%乙腈、0.05~0.2wt%甲酸水溶液补足100%组成;流速0.2~0.4ml/min;柱温:35~45℃;进样量4~6μl。
12、优选的,所述步骤3中质谱扫描范围m/z 100~1500da,雾化气压50~60psi,辅助气压50~60psi,气帘气压30~40psi。
13、优选的,所述步骤3中正离子模式下参数为:毛细管电压5000~6000v,裂解电压80~120v,碰撞能量30~40ev。
14、优选的,所述步骤3中负离子模式下参数为:毛细管电压-4000~-5000kv,裂解电压-70~-90v,碰撞能量30~40ev。
15、所述改性固定相的制备方法如下,以重量份计:
16、s1、将4~6份二氧化硅粉末、8~12份3-(异丁烯酰氧)丙基三甲氧基硅烷、8~12份水和0.5~2份乙酸加入到18~22份无水乙醇中,在50~70℃油浴中搅拌20~40min,静置3~5h,得到预处理物;
17、s2、将0.1~0.3份甲基丙烯酰胺、0.04~0.06份海藻酸钠和0.04~0.06份n,n'-亚甲基-双丙烯酰胺加入到30~50份水中,进行超声处理,然后加入2~4份步骤s1制备的预处理物,100~500rpm磁力搅拌0.5~2h,静置8~15h,再加入0.04~0.06份过硫酸铵、0.004~0.006份硫酸钙和0.1~0.2份金属有机骨架材料,在50~70℃油浴中进行交联聚合1~3h,用氮气鼓泡除去混合物中溶解的空气,然后加热至50~70℃,100~300rpm搅拌回流冷凝4~6h,随后,温度加热到70~90℃,在100~200rpm搅拌下完全蒸发溶剂,然后,加入40~60份水浸泡一夜,最后用水洗涤1~3次,在70~90℃真空烘箱中干燥5~15h,得到后处理物;
18、s3、将2~3份后处理物在20~30份四氯甲烷中超声处理,以正己烷为推进液,在30~50mpa的压力下将混合物填充到140~160×4~5mm不锈钢管中,当流出的液体体积超过100ml时,床层稳定,充填完成,得到改性固定相。
19、优选的,所述超声处理时间各自独立地为10~30min、超声功率各自独立地为200~500w、超声频率各自独立地为20~60khz。
20、本发明采用uplc-q-tof-ms/ms技术对甘草多糖化学成分进行定性分析,以明确甘草多糖中化学成分。
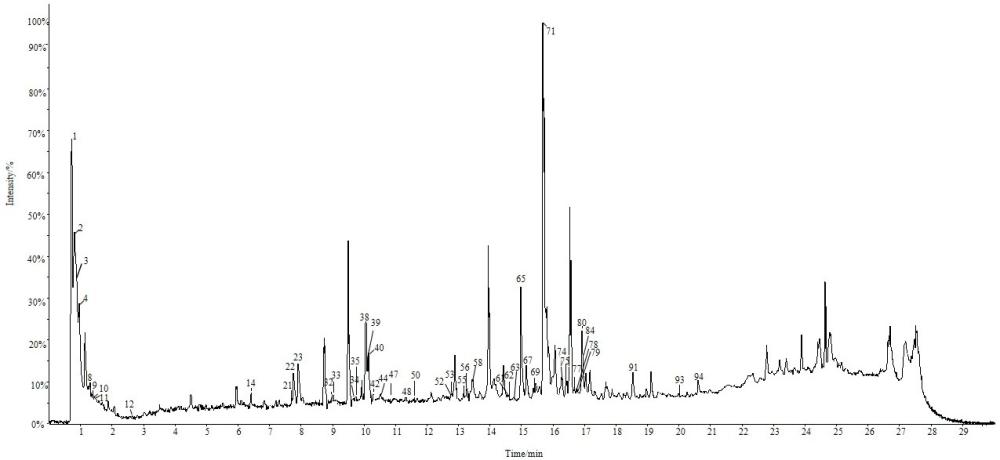
21、采用waters acquity uplc beh-c18色谱柱(2.1 mm×100 mm,1.7 μm);乙腈-甲酸水溶液为流动相进行梯度洗脱,流速0.3 ml/min,柱温40℃,进样量5μl,固定相为改性固定相。采用电喷雾离子源,在正、负离子模式下对甘草多糖进行质谱扫描。通过analyst tf1.8.1软件和sciex os 2.0.0软件进行数据采集和数据处理。根据高分辨质谱提供的化合物相对分子质量、离子碎片信息,参考文献报道的质谱数据并对比数据库分析结果,对甘草多糖中化学成分进行分析。
22、在正、负离子模式下共分析出95种化学成分,其中包括57种黄酮类成分,18种三萜类成分,4种香豆素类成分,3种有机酸类成分,其他类包括7种氨基酸类成分、3种糖类成分、3种核苷类成分,共13种。uplc-q-tof-ms/ms技术能有效分析出甘草多糖中化学成分,可为甘草多糖后续实验提供支撑。
23、与现有技术相比,本发明的有益效果:
24、1)本发明通过使用uplc-q-tof-ms/ms技术能有效分析出甘草多糖中化学成分,可为甘草多糖后续实验提供支撑。
25、2)本发明采用的改性固定相中存在多模式相互作用,具有更好的分离性能和良好的稳定重复性,在实际应用中具有重要作用,拓展了在色谱分离领域的潜在应用。
- 还没有人留言评论。精彩留言会获得点赞!