分离测定克霉唑中间体Z1中有关物质的高效液相色谱法的制作方法
本发明属于药物化学分析,具体涉及一种分离测定克霉唑中间体z1中有关物质的高效液相色谱法。
背景技术:
1、克霉唑(clotrimazole)属于吡咯类广谱抗真菌药,主要用于治疗敏感菌所致的深部和浅部真菌病,如隐球菌脑膜炎、念珠菌肺炎、肠炎、组织胞浆病、体癣、手足癣等。克霉唑对多种真菌尤其是白色念珠菌具有较好抗菌作用,其作用机制是抑制真菌细胞膜的合成,以及影响其代谢过程。克霉唑中间体z1(clr-z1)为克霉唑合成过程中的关键中间体,其中文化学名为1-氯-2-(氯二苯基甲基)苯,分子式为c19h14cl2,分子量为313.22,cas号为42074-68-0,结构式如式1所示。
2、
3、在研究克霉唑中间体z1的过程中发现,可能存在如下杂质:杂质sm1、杂质z1a、杂质z1b、杂质z1c、杂质z1d、杂质z1e、杂质z1m、杂质z1n、杂质z1i、杂质z1j、杂质z1f、杂质z1g。其中,杂质sm1为关键的起始原料,杂质z1a和杂质z1d则为工艺中产生可能性非常大的杂质,故需定入标准。此外,克霉唑中间体z1、杂质z1a、杂质z1d、杂质z1m、杂质z1i均极易水解,水解为杂质z1c、杂质z1b、杂质z1e、杂质z1n、杂质z1j,这为克霉唑中间体z1中相关杂质的控制增加了一定难度。所以,为保障克霉唑原料药及产品的质量,有必要对克霉唑中间体z1中上述有关杂质进行研究和控制。
4、目前,暂无文献或专利资料对克霉唑中间体z1中上述有关物质的测定方法进行报道,因此,有必要建立新的方法来实现克霉唑中间体z1中有关物质的控制,以保障克霉唑原料药和制剂的质量。
技术实现思路
1、有鉴于此,本发明的目的之一在于提供一种采用高效液相色谱法对克霉唑中间体z1中有关物质进行分离的方法,该方法专属性强,灵敏度高,为后续克霉唑中间体z1中有关物质的定性和定量检测提供了支持。
2、为实现上述目的,本发明采用以下技术方案:
3、采用高效液相色谱法对克霉唑中间体z1中有关物质进行分离的方法,所述有关物质为杂质sm1、杂质z1a、杂质z1b、杂质z1c、杂质z1d、杂质z1e、杂质z1m、杂质z1n、杂质z1i、杂质z1j、杂质z1f和杂质z1g中的任一种或多种;所述高效液相色谱法为:采用十八烷基硅烷键合硅胶为色谱柱填充剂,以磷酸水溶液为流动相a,甲醇为流动相b,通过线性梯度洗脱将克霉唑中间体z1中所述有关物质进行分离;
4、所述克霉唑中间体z1的结构式如式1所示;所述杂质sm1的结构式如式2所示,所述杂质z1a的结构式如式3所示,所述杂质z1b的结构式如式4所示,所述杂质z1c的结构式如式5所示,所述杂质z1d的结构式如式6所示,所述杂质z1e的结构式如式7所示,所述杂质z1m的结构式如式8所示,所述杂质z1n的结构式如式9所示,所述杂质z1i的结构式如式10所示,所述杂质z1j的结构式如式11所示,所述杂质z1f的结构式如式12所示,所述杂质z1g的结构式如式13所示;
5、
6、
7、进一步,程序运行时间为48分钟。
8、进一步,样品洗脱顺序为:杂质z1a和/或杂质z1b、杂质z1d和/或杂质z1e、克霉唑中间体z1和/或杂质z1c、杂质z1m和/或杂质z1n、杂质sm1、杂质z1i和/或杂质z1j、杂质z1f、杂质z1g。可以根据洗脱先后对各组分进行定性。
9、进一步,所述梯度洗脱设置程序如下:
10、0分钟,设置所述流动相a和所述流动相b的体积比为35-45:65-55;
11、3分钟,设置所述流动相a和所述流动相b的体积比为35-45:65-55;
12、35分钟,设置所述流动相a和所述流动相b的体积比为5-15:95-85;
13、37分钟,设置所述流动相a和所述流动相b的体积比为5-15:95-85;
14、38分钟,设置所述流动相a和所述流动相b的体积比为35-45:65-55;
15、48分钟,设置所述流动相a和所述流动相b的体积比为35-45:65-55。
16、作为优选,设置所述梯度洗脱的程序如下:
17、0分钟,设置所述流动相a和所述流动相b的体积比为38-42:62-58;
18、3分钟,设置所述流动相a和所述流动相b的体积比为38-42:62-58;
19、35分钟,设置所述流动相a和所述流动相b的体积比为10:90;
20、37分钟,设置所述流动相a和所述流动相b的体积比为10:90;
21、38分钟,设置所述流动相a和所述流动相b的体积比为38-42:62-58;
22、48分钟,设置所述流动相a和所述流动相b的体积比为38-42:62-58。
23、作为最优选,设置所述梯度洗脱的程序如下:
24、0分钟,设置所述流动相a和所述流动相b的体积比为40:60;
25、3分钟,设置所述流动相a和所述流动相b的体积比为40:60;
26、35分钟,设置所述流动相a和所述流动相b的体积比为10:90;
27、37分钟,设置所述流动相a和所述流动相b的体积比为10:90;
28、38分钟,设置所述流动相a和所述流动相b的体积比为40:60;
29、48分钟,设置所述流动相a和所述流动相b的体积比为40:60。
30、进一步,所述流动相a的浓度为0.07%~0.13%。
31、作为优选,所述流动相a的浓度为0.09%~0.11%,更优选为0.1%。
32、进一步,所述流动相的流速为0.7-1.3ml/min;所述色谱柱柱箱温度为17-23℃。
33、作为优选,所述流动相的流速为0.9-1.1ml/min,更优选为1.0ml/min;所述色谱柱柱箱温度为18-22℃,更优选为20℃。
34、进一步,进样体积为10μl。
35、本发明的目的之二在于提供一种鉴别克霉唑中间体z1中是否含有有关物质的方法,该方法可以实现克霉唑中间体z1中有关物质的有效鉴别。
36、为实现上述目的,本发明采用以下技术方案:
37、鉴别克霉唑中间体z1中是否含有有关物质的方法,采用前述分离方法对克霉唑中间体z1中有关物质进行分离;进入检测器进行检测,得到色谱图;所述检测器的检测波长为220-230nm。
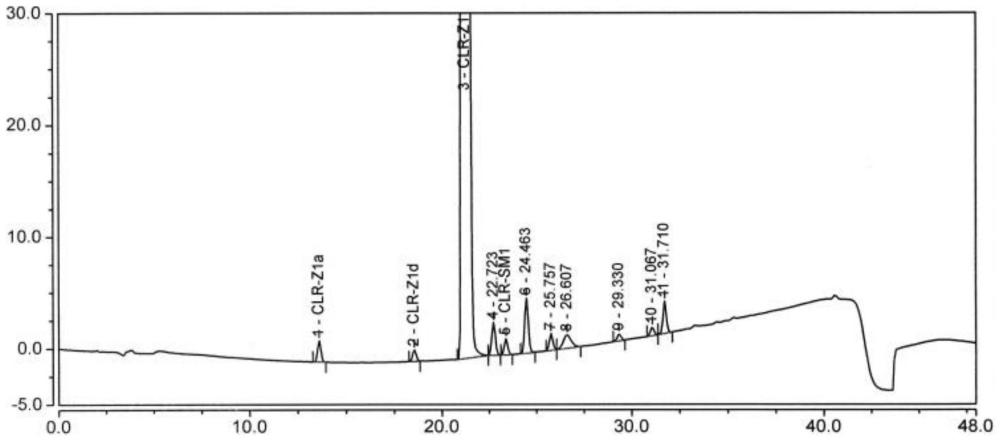
38、进一步,各组分保留时间由短到长依次为:杂质z1a和/或杂质z1b、杂质z1d和/或杂质z1e、克霉唑中间体z1和/或杂质z1c、杂质z1m和/或杂质z1n、杂质sm1、杂质z1i和/或杂质z1j、杂质z1f、杂质z1g。
39、可以根据保留时间对各组分进行定性。
40、进一步,色谱柱规格为4.6mm×250mm,5μm;流动相a为浓度为0.1%的磷酸水溶液,流动相b为甲醇;流速为1.0ml/min;柱度为20℃;检测波长为220-230nm;
41、按如下梯度洗脱程序进行线性梯度洗脱,并得到色谱图;
42、0分钟,设置所述流动相a和所述流动相b的体积比为40:60;
43、3分钟,设置所述流动相a和所述流动相b的体积比为40:60;
44、35分钟,设置所述流动相a和所述流动相b的体积比为10:90;
45、37分钟,设置所述流动相a和所述流动相b的体积比为10:90;
46、38分钟,设置所述流动相a和所述流动相b的体积比为40:60;
47、48分钟,设置所述流动相a和所述流动相b的体积比为40:60。
48、进一步,保留时间在13.6±0.5min,判定为杂质z1a和/或杂质z1b;保留时间在18.5±0.5min,判定为杂质z1d和/或杂质z1e;保留时间在21.3±0.5min,判定为克霉唑中间体z1和/或杂质z1c;保留时间在22.7±0.5min,判定为杂质z1m和/或杂质z1n;保留时间在23.4±0.5min,判定为杂质sm1;保留时间在24.5±0.5min,判定为杂质z1i和/或杂质z1j;保留时间在25.8±0.5min,判定为杂质z1f;保留时间在31.7±0.5min,判定为杂质z1g。
49、本发明的目的之三在于提供一种检测克霉唑中间体z1中有关物质的方法含量的方法,该方法可在48分钟内或大于48分钟的时间实现克霉唑中间体z1中多种有关物质的含量检测,为进一步提高克霉唑原料药或制剂的质量提供了新的思路。
50、为实现上述目的,本发明采用以下技术方案:
51、检测克霉唑中间体z1中有关物质的方法含量的方法,包括如下步骤:
52、(1)采用前述方法对克霉唑中间体z1中有关物质进行分离和检测,得到色谱图;
53、(2)根据步骤(1)得到的色谱图,采用外标法以峰面积计算杂质z1a和/或杂质z1b、杂质z1d和/或杂质z1e、杂质sm1的含量;采用主成分自身对照法计算杂质z1m和/或杂质z1n、杂质z1i和/或杂质z1j、杂质z1f、杂质z1g的含量。
54、进一步,杂质计算公式如下:
55、1)外标法计算有关物质:
56、
57、
58、式中,ar:对照品溶液中杂质峰面积;
59、mr:对照品重量;
60、nr:对照品稀释倍数;
61、p:对照品含量;
62、as:供试品溶液中杂质峰面积;
63、ms:供试品重量;
64、ns:供试品稀释倍数。
65、2)加校正因子自身对照法计算有关物质:
66、
67、式中,az:对照溶液中主成分峰面积;
68、as:供试品溶液中杂质峰面积;
69、f:杂质校正因子(没有校正因子的,按f=1计);
70、n:配制对照溶液的稀释倍数。
71、进一步,样品配制溶剂为乙腈水溶液,所述乙腈水溶液中,乙腈和水的体积比为70:30。
72、作为优选,供试品溶液浓度为1mg/ml。
73、作为优选的技术方案:
74、步骤1.精密称定本品,如25mg,置25ml量瓶中,加溶剂超声溶解并稀释至刻度,摇匀,作为供试品/样品溶液;
75、步骤2.精密量取步骤1配制的供试品溶液,如1.0ml,置50ml量瓶中,用溶剂稀释至刻度,摇匀;精密量取1.0ml,置10ml量瓶中,用溶剂稀释至刻度,摇匀,作为对照溶液;
76、步骤3.取杂质sm1、杂质z1b(杂质z1b转换为杂质z1a,乘以系数1.2534)与杂质z1e(杂质z1e转换为杂质z1d,乘以系数1.0709)各适量,精密称定,加溶剂溶解并稀释制成每1ml约含杂质sm1 1μg、杂质z1a 2μg与杂质z1d 1μg的混合溶液,作为对照品溶液;
77、步骤4.精密量取供试品溶液、对照品溶液与对照溶液,分别注入高效液相色谱仪进行检测,得到色谱图;根据测得的色谱图,采用外标法以峰面积计算杂质z1a和/或杂质z1b、杂质z1d和/或杂质z1e、杂质sm1的含量;采用主成分自身对照法计算杂质z1m和/或杂质z1n、杂质z1i和/或杂质z1j、杂质z1f、杂质z1g的含量。
78、本发明将杂质水解后进样,建立相应的hplc方法。杂质z1a与杂质z1b合并控制,杂质z1d与杂质z1e合并控制,选择分子量大响应值更低的杂质z1a和z1d用于定量检测。由于杂质z1a和杂质z1d易水解不稳定,用z1b转换为z1a,乘以转换系数1.2534配制对照品溶液,z1e转换为z1d,乘以转换系数1.0709配制对照品溶液,外标法计算,严格控制质量水平。杂质z1m+z1n、杂质z1i+z1j、杂质z1f、杂质z1g作为其他单杂控制。杂质z1c则另建方法研究。
79、本发明的有益效果在于:
80、本发明提出的高效液相色谱法,可以将克霉唑中间体z1中可能存在的多种有关物质进行分离和检测,包括杂质sm1、杂质z1a、杂质z1b、杂质z1c、杂质z1d、杂质z1e、杂质z1m、杂质z1n、杂质z1i、杂质z1j、杂质z1f和/或杂质z1g。本发明方法具有操作简单、专属性强、灵敏度高、耐用性好、准确度高等优点。本发明方法为克霉唑中间体z1中有关物质的定性和定量提供了新的方法,为克霉唑的质量控制提供了新的思路。
- 还没有人留言评论。精彩留言会获得点赞!