当归补血颗粒基准样品的指纹图谱检测方法及其指纹图谱与流程
本发明属于中药检测,具体涉及当归补血颗粒基准样品的指纹图谱检测方法及其指纹图谱。
背景技术:
1、当归补血汤为国家中医药管理局2018年4月发布的《古代经典名方目录(第一批)》中的第51首处方,出自金•李东垣《内外伤辩惑论》,由黄芪、当归按5:1的比例配伍而成,具有益气生血的功效,主治血虚阳浮发热证,为甘温除热法代表方之一。
2、经典名方基准样品是指以古代医籍中记载古代经典名方制备方法为依据制备而得的中药药用物质,除成型工艺外,其余制备方法与古代医籍记载基本一致。基准样品作为经典名方制剂药用物质确定基准,是桥接经典名方传统制法与现代工业生产工艺的对照物质。经典名方复方制剂的开发是以出膏率、浸出物、指纹图谱、含量测定等指标为关键质量属性,其中指纹图谱及含量测定是中药复方制剂质量控制最重要的手段。通过研究各指标成分从药材、饮片到中间体、制剂的转移率,以及各特征峰的传递情况,并与基准样品进行质量对比,可较全面的说明生产全过程的量值传递情况,因此研究开发基准样品指纹图谱及含量测定方法具有重要意义。
3、目前对当归补血汤基准样品及其复方制剂的研究中,多为单独控制其特征图谱或含量测定,且现有技术中的特征图谱方法分析时间较长,部分特征峰分离度不够、峰型较差,另外,含量测定指标多为阿魏酸和毛蕊异黄酮葡萄糖苷,缺少对挥发油成分的定量控制,无法全面控制当归补血颗粒基准样品质量。
4、公开专利cn103381217a提出了一种六味补血胶囊及其质量控制方法与应用,采用高效液相法,鉴别六味补血胶囊中是否含有白芍、当归、黄芪、熟地黄、紫草、川芎及其成分,测定六味补血胶囊的体外溶出行为,以指纹图谱鉴别有效成分群;以毛蕊花糖苷与毛蕊异黄酮葡萄糖苷为对照品同时测定六味补血胶囊中含量;采用气相色谱法测定六味补血胶囊中挥发性成分藁本内酯的含量,色谱条件为十八烷基硅烷键合硅胶为填充剂的色谱柱,乙腈-0.35%乙酸溶液(25:75)为流动相;紫外检测器;波长为270nm,流速为1ml/min,理论板数以毛蕊花糖苷峰计算应不低于2000,按照此方法所提供指纹图谱的建立方法,依据10批或10批以上供试品所测得的图谱,制定标准图谱。所得六味补血制剂的标准指纹图谱中有10个特征共有峰,其中2号色谱峰为芍药苷参照峰,3号色谱峰为阿魏酸参照峰,5号色谱峰为丹皮酚参照峰,8号色谱峰为川芎嗪参照峰,最强峰为3号色谱峰,图谱总长度为60min。但所述指纹图谱方法采用普通液相色谱仪,分析时间较长,且单独建立藁本内酯方法,增加了检验时间及成本。
技术实现思路
1、本发明要解决的技术问题是克服现有技术存在的上述缺陷,提供当归补血颗粒基准样品的指纹图谱检测方法,极大地缩短了分析时间,提高检验效率,又能同时控制方中两味药材的多个含量测定指标,能全面控制经典名方当归补血颗粒基准样品质量。
2、本发明还提供当归补血颗粒基准样品的指纹图谱。
3、本发明所述的当归补血颗粒基准样品的指纹图谱检测方法:包括以下步骤:
4、(1)参照物溶液的制备:取毛蕊异黄酮葡萄糖苷、阿魏酸、藁本内酯对照品,混合,加溶剂溶解,配制成参照物溶液;
5、供试品溶液的制备:以当归补血颗粒水煎液直接冻干作为基准样品,称取基准样品,加溶剂提取,过滤,取续滤液,即得;
6、单味饮片阴性对照溶液的制备:以黄芪阴性样品、酒当归阴性样品的单味饮片的水煎液直接冻干后,加溶剂提取,过滤,取续滤液,即得;
7、(2)分别吸取参照物溶液、供试品溶液及单味饮片阴性对照溶液注入超高效液相色谱仪,进行测定,按色谱条件进样,得到参照物溶液、供试品溶液及单味饮片阴性对照溶液的液相色谱图;
8、所述的色谱条件包括:以乙腈为流动相a,体积百分比0.01%~0.05%甲酸溶液为流动相b,进行梯度洗脱;
9、(3)标准指纹图谱的建立:采用国家药典委员会制订的《中药色谱指纹图谱相似度评价系统》,对多批次供试品溶液色谱数据进行匹配,即得标准指纹图谱;
10、(4)含量的测定:以步骤(1)中的参照物溶液的制备方法制备混合对照品溶液:以步骤(1)中的供试品溶液的制备方法制备待测供试品溶液,分别吸取混合对照品溶液、待测供试品溶液,注入超高效液相色谱仪,按照步骤(2)的色谱条件,进行当归补血颗粒基准样品中的毛蕊异黄酮葡萄糖苷、藁本内酯、阿魏酸含量的测定。
11、步骤(2)所述的色谱条件还包括:色谱柱:以十八烷基硅烷键合硅胶为填充剂;
12、流速:0.35~0.45ml/min;
13、柱温:25~35℃;
14、检测波长:240~300nm;
15、进样量:2~5μl;
16、理论板数按毛蕊异黄酮葡萄糖苷峰计算不低于10000;
17、梯度洗脱条件:0~1.5min,流动相a的体积分数变化为10%~10%,流动相b的体积分数变化为90%~90%;1.5~4min,流动相a的体积分数变化为10%~20%,流动相b的体积分数变化为90%~80%;4~5.5min,流动相a的体积分数变化为20%~25%,流动相b的体积分数变化为80%~75%;5.5~6.5min,流动相a的体积分数变化为25%~25%,流动相b的体积分数变化为75%~75%;6.5~9min,流动相a的体积分数变化为25%~55%,流动相b的体积分数变化为75%~45%;9~10min,流动相a的体积分数变化为55%~90%,流动相b的体积分数变化为45%~10%;10~11min,流动相a的体积分数变化为90%~55%,流动相b的体积分数变化为10%~45%;11~12min,流动相a的体积分数变化为55%~45%,流动相b的体积分数变化为45%~55%;12~13min,流动相a的体积分数变化为45%~10%,流动相b的体积分数变化为55%~90%;13~18min,流动相a的体积分数变化为10%~10%,流动相b的体积分数变化为90%~90%。
18、色谱柱为thermo scientific accucore色谱柱,2.1×150mm,2.6μm。
19、所述的当归补血颗粒包括君药黄芪、臣药酒当归。
20、步骤(1)所述的溶剂为质量含量50%甲醇。
21、步骤(1)所述的提取方式为超声或回流,优选超声。步骤(1)所述的提取时间为20~40min,优选30min。
22、步骤(3)的多批次供试品溶液为18批次,18批当归补血颗粒基准样品指纹图谱的相似度在0.933~0.997之间,均大于0.90。
23、步骤(3)所检测的当归补血颗粒基准样品中的毛蕊异黄酮葡萄糖苷的含量范围为0.47mg/g~1.05mg/g;阿魏酸的含量范围为0.24mg/g~0.58mg/g;藁本内酯的含量范围为0.27mg/g~0.78mg/g。
24、一种当归补血颗粒基准样品的指纹图谱,是所述的当归补血颗粒基准样品的指纹图谱检测方法中的步骤(3)的标准指纹图谱。
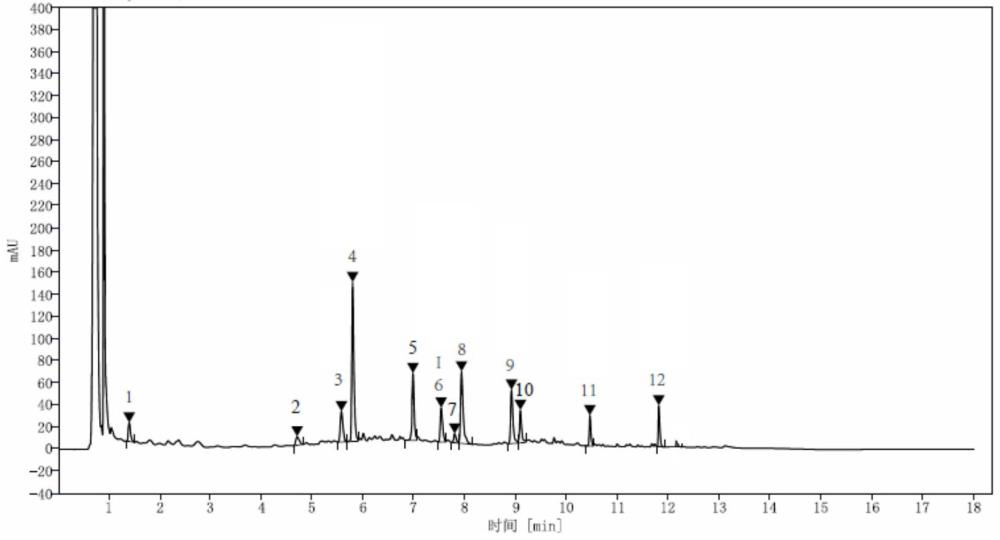
25、呈现12个特征峰,其中峰3为阿魏酸峰,峰4为毛蕊异黄酮葡萄糖苷峰,峰6为洋川芎内酯i峰,峰8为芒柄花苷峰,峰9为毛蕊异黄酮峰,峰11为芒柄花素峰,峰12为藁本内酯峰。本发明采用超高效液相色谱仪,既缩短了检测时间,又能同时控制包括藁本内酯在内的3个含量测定指标,极大地提高了检验效率,降低了检验成本。
26、具体的,所述的当归补血颗粒基准样品的指纹图谱检测方法:包括以下步骤:
27、(1)参照物溶液的制备:取毛蕊异黄酮葡萄糖苷、阿魏酸、藁本内酯对照品,精密称定,置棕色量瓶中混合,加50%甲醇溶剂溶解,配制成1ml分别含50μg毛蕊异黄酮葡萄糖苷、12μg阿魏酸、10μg藁本内酯的混合溶液;
28、供试品溶液的制备:以当归补血颗粒水煎液直接冻干作为基准样品,称取基准样品1g,精密称定,置具塞锥形瓶中,加50%甲醇溶剂25ml进行超声或回流提取30min,放冷,再称定重量,用50%甲醇补足减失的重量,摇匀,静置,取上清液,过滤,取续滤液,即得;
29、单味饮片阴性对照溶液的制备:取黄芪阴性样品0.17g、酒当归阴性样品0.83g的单味饮片的水煎液直接冻干后,分别精密称定,置具塞锥形瓶中,加50%甲醇溶剂25ml进行超声或回流提取30min,放冷,再称定重量,用50%甲醇补足减失的重量,摇匀,静置,取上清液,过滤,取续滤液,即得。
30、(2)分别吸取参照物溶液、供试品溶液及单味饮片阴性对照溶液注入超高效液相色谱仪,进行测定,按色谱条件进样,得到参照物溶液、供试品溶液及单味饮片阴性对照溶液的液相色谱图;
31、所述的色谱条件包括:
32、色谱柱:以十八烷基硅烷键合硅胶为填充剂,thermo scientific accucore色谱柱,2.1×150mm,2.6μm;
33、流速:0.4ml/min;
34、柱温:30℃;
35、检测波长:240~300nm;
36、进样量:2~5μl;
37、理论板数按毛蕊异黄酮葡萄糖苷峰计算不低于10000;
38、流动相:以乙腈为流动相a,以体积百分比0.03%甲酸溶液为流动相b,进行梯度洗脱;
39、梯度洗脱条件如表1所示,流动相中的%为体积百分比。
40、表1洗脱条件
41、
42、(3)标准指纹图谱的建立:取供试品溶液及单味饮片阴性对照溶液,按色谱条件进样,记录色谱图,得到全方与单味饮片阴性对照对比图,采用对照品进行特征峰指认,取各对照品溶液,按色谱条件进样,记录色谱图,取18批当归补血颗粒基准样品作为供试品溶液,得到的色谱数据,采用国家药典委员会制订的《中药色谱指纹图谱相似度评价系统》(2012版),以s1为参照谱图,选择相对稳定、响应值较高、峰型良好的特征峰进行多点校正,并计算相似度,相似度在0.933~0.997之间,得到标准指纹图谱,指纹图谱中具有12个特征峰,其中峰3为阿魏酸峰,峰4为毛蕊异黄酮葡萄糖苷峰,峰6为洋川芎内酯i峰,峰8为芒柄花苷峰,峰9为毛蕊异黄酮峰,峰11为芒柄花素峰,峰12为藁本内酯峰。
43、(4)含量的测定:按照步骤(1)制备出毛蕊异黄酮葡萄糖苷、阿魏酸、藁本内酯对照品溶液:以步骤(1)中的供试品溶液的制备方法制备待测供试品溶液,分别吸取混合对照品溶液、待测供试品溶液,注入超高效液相色谱仪,按照步骤(2)的色谱条件,进行当归补血颗粒基准样品中的毛蕊异黄酮葡萄糖苷、阿魏酸、藁本内酯的线性关系,毛蕊异黄酮葡萄糖苷的线性关系为y=18.3008x+2.0126,阿魏酸的线性关系为y=10.2492x-2.1722,藁本内酯的线性关系为y=5.6332x-0.0931,根据线性关系,进一步检测出当归补血颗粒中的毛蕊异黄酮葡萄糖苷、阿魏酸、藁本内酯含量。
44、与现有技术相比,本发明具有的有益效果是:
45、(1)本发明在同一色谱条件下,既测定出了当归补血颗粒基准样品的指纹图谱,同时又测定了方中君药黄芪中的活性成分毛蕊异黄酮葡萄糖苷(黄酮类)和臣药酒当归中的活性成分阿魏酸(有机酸类)、藁本内酯(挥发油类)的含量,将定性与定量相结合,缩短了检测时间和成本,提高了工作效率,更加全面的控制了当归补血颗粒基准样品的质量,进而控制了当归补血颗粒的质量。
46、(2)本发明建立的标准指纹图谱比常规的特征图谱更加严格,进一步提升了当归补血颗粒基准样品的质量标准。
47、(3)本发明控制了挥发油中藁本内酯的含量,填补了检测领域的空白。
48、(4)本发明的方法专属性好、精密度高、重现性好、准确度高、耐用性良好,样品处理简单快捷。
- 还没有人留言评论。精彩留言会获得点赞!