寻找样本的染色体突变位点的分析方法和分析装置与流程
本发明涉及生物和计算机领域,特别涉及一种通过计算机寻找样本的染色体突变位点的分析方法和分析装置。
背景技术:
基于高通量测序数据寻找和致病基因、癌症治疗、个性化用药相关的染色体突变位点为临床应用提供了不可估量的前景,例如:癌症基因组、无创产前诊断、药物靶标筛选。高通量测序技术能否在这些领域取得进展,其关键在于分析方法及软件的创新。
由于测序技术的不断进步,测序价格的不断降低,我们得到的数据越来越多,如何快速的挖掘出需要的信息已经是亟待解决的问题。目前可以进行同样分析的软件主要有Samtools、GATK等。而这些软件是单线程,对于大量数据的分析是很慢的,因此急需寻找一个新的方式来缩短分析的时间。
技术实现要素:
有鉴于此,本发明基于寻找基因突变的理论和分布式计算框架提供了一种分析速度更快的数据的软件分析方法和分析装置。
本发明的实施例提供了一种基于高通量测序数据寻找样本的染色体突变位点的分析方法,所述方法包括:
根据测序序列和参考序列的比对结果为并行计算准备数据;
通过并行计算过滤掉准备好的数据中无效的碱基;
根据过滤后保留的碱基确定样本的染色体突变位点。
所述测序序列和参考序列的比对结果为并行计算准备数据包括:
对包含所述比对结果的数据进行切分;
生成包含切分后的数据的执行实体。
优选地,所述对包含所述比对结果的数据进行切分包括:
将所述比对结果的数据的文件切分为文件块;
每隔预定时间间隔将从所述文件块中读取到的数据切分为组成DStream的RDD。
优选地,所述生成包含切分后的数据的执行实体包括根据RDD生成执行实体job。
所述通过并行计算过滤掉准备好的数据中无效的碱基包括:
通过执行实体对准备好的数据进行并行计算:当由样本产生的测序序列与参考序列对应位点上的碱基不一致时,对此测序序列上的每个碱基计算碱基比对质量值;
通过执行实体并行地在所述碱基比对质量值和此碱基的测序质量值中取较小值作为此碱基的最终质量值;
通过执行实体并行地进行过滤:如果所述最终质量值小于此碱基的第一阈值,则过滤掉此碱基。
所述根据过滤后保留的碱基确定样本的染色体突变位点包括:
根据过滤后保留的碱基确定样本的每个碱基位点上的突变碱基所占的比率;
如果所述样本的某一位点上的突变碱基所占的比率大于等于第二阈值,则此位点为样本的染色体突变位点。
本发明的实施例还提供了一种基于高通量测序数据寻找样本的染色体突变位点的分析装置,所述装置包括:
并行准备模块,用于根据测序序列和参考序列的比对结果为并行计算准备数据;
并行计算模块,用于通过并行计算过滤掉准备好的数据中无效的碱基;
结果合并模块,用于根据过滤后保留的碱基确定样本的染色体突变位点。
优选地,所述并行准备模块包括:
数据切分单元,用于对包含所述比对结果的数据进行切分;
执行实体生成单元,用于生成包含切分后的数据的执行实体。
所述数据切分单元,用于:
将包含所述比对结果的数据的文件切分为文件块;
每隔预定时间间隔将从所述文件块中读取到的数据切分为组成DStream的RDD。
所述执行实体生成单元用于根据RDD生成执行实体job。
优选地,所述并行计算模块包括:
质量计算单元,用于:
通过执行实体对准备好的数据进行并行计算:当由样本产生的测序序列与参考序列对应位点上的碱基不一致时,对此测序序列上的每个碱基计算碱基比对质量值,
通过执行实体并行地在所述碱基比对质量值和此碱基的测序质量值中取较小值作为最终质量值;
过滤单元:用于通过执行实体并行地进行过滤:当所述最终质量值小于此碱基的第一阈值时,过滤掉此碱基。
优选地,所述结果合并模块包括:
比率计算单元,用于根据过滤后保留的碱基确定样本的每个碱基位点上的突变碱基所占的比率;
突变位点确定单元,用于当所述样本的某一位点上的突变碱基所占的比率大于等于第二阈值时,确定此位点为样本的染色体突变位点。
采用了本发明的技术方案以后,可以采用多机器弹性并行计算来加快分析速度。Spark采用内存迭代计算,从缓存中存入和读取的时间很短,几乎可以忽略,而另一种可选方案需要向HDFS读写数据则需要耗费大量时间。本发明的技术方案与其他软件的解决方案相比,分析速度大幅提升。
附图说明
图1为本发明第一实施例提供的基于高通量测序数据寻找样本的染色体突变位点的分析方法的示意图;
图2为本发明第二实施例提供的基于高通量测序数据寻找样本的染色体突变位点的分析方法的示意图;
图3为本发明第三实施例提供的寻找样本的染色体突变位点的分析方法的示意图;
图4为隐马尔可夫模型(HMM)的示意图;
图5为本发明实施例提供的基于高通量测序数据寻找样本的染色体突变位点的分析装置的结构框图。
具体实施方式
本发明提供了一种基于高通量测序数据寻找样本的染色体突变位点的分析方法和分析装置。下面三个实施例对本发明进行说明。
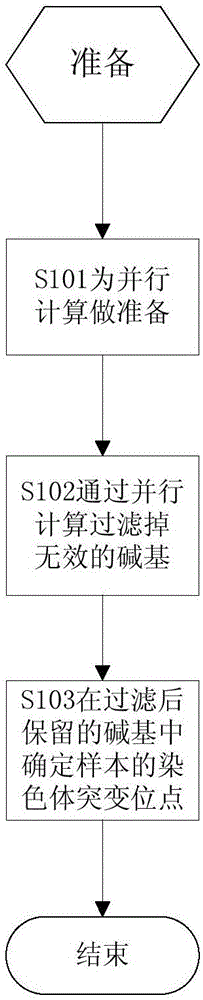
请参阅图1,在本发明第一实施例中,首先根据碱基比对结果为并行计算进行准备,然后通过并行计算在准备好的数据中过滤掉无效的碱基,最后根据过滤后保留的碱基确定样本的染色体突变位点。
S101、根据碱基比对结果为并行计算准备数据。
将包含测序序列和参考序列碱基比对结果的文件切分成多个文件块,保存在文件系统中。
S102、通过并行计算过滤掉准备好的数据中无效的碱基。
多台计算机同时从文件系统中的多个文件块中读取数据,通过并行计算过滤掉其中无效的碱基。
S103、根据过滤后保留的碱基确定样本的染色体突变位点。
请参阅图2,在本发明的第二实施例中,通过使用hadoop框架进行并行计算寻找样本的染色体突变位点:首先将包含碱基比对结果的大文件切分为多个小数据块保存到HDFS中,再利用多台计算机并行地从所述多个小数据块中读取数据,通过并行计算过滤掉无效的碱基。将多台计算机上并行计算的结果合并,确定样本的染色体突变位点。
具体地,在本发明的第二实施例中:
第一阈值是指当测序序列某一碱基的最终质量值小于此值时,认为此碱基无效,并将其过滤掉,不作为寻找样本的染色体突变位点的依据;
第二阈值是指对于样本的某一确定位点,如果其突变碱基所占的比率大于等于此值,则确定此位点为样本的染色体突变位点。
S201、将包含碱基比对结果的大数据文件切分为多个小数据块。
从数据源输入的文件为bam或者sam文件,其中包含了测序序列与参考序列碱基的比对结果的大量数据。
多个客户端通过网络将原始的bam或sam大文件上传到HDFS(HadoopDistributed File System),HDFS将此大文件根据一定的规则切分成小数据块并保存。切分规则如下:比如输入的大文件为3G,在spark streaming中将一个block设置为128M,那么输入的大文件总共将被切分为3*1024/128=24块。
S202、Hadoop读取HDFS中的小数据块,启动job,job为每一个小文件块生成实际的执行实体map task和reduce task。
参与并行计算的多台计算机上的Hadoop并行地读取HDFS中的小数据块,并启动一个job,job为每一个小数据块生成一个map task和一个reduce task。Map task和reduce task是并行计算的执行实体。
S203、在map task中,通过并行计算碱基比对质量值(BAQ)和最终质量值来过滤掉测序序列位点上无效的碱基,并将过滤后保留下来的碱基保存到HDFS中。
Hadoop在参与并行计算的多台计算机上并行地执行步骤S202中生成的map task和reduce task,先运行map task,然后运行reduce task。
为了寻找样本的染色体突变位点,需要通过计算碱基比对质量值(BAQ)和最终质量值过滤掉测序序列上的无效碱基。这一部分并行计算是在map task中进行的。有关计算碱基比对质量值BAQ和最终质量值以及过滤掉测序序列位点上无效碱基的算法描述请参见本说明书步骤S303至S306的相关描述。
S204、在reduce task中,根据保留下来的碱基通过并行计算确定样本的每一个位点上的突变碱基所占的比率,并根据此比率确定样本的染色体突变位点。
在reduce task中,首先从HDFS中读取通过步骤S203过滤后保留下来的碱基。然后,对于样本上的每一个位点,累加由测序数据产生的ATCG各碱基的次数(通过多台计算机对其上的小数据块进行并行计算得出),从而得到此位点上的突变碱基所占的比率。
如果样本某一位点上的突变碱基所占的比率大于等于第二阈值,则确定此位点是样本的染色体突变位点;否则确定此位点不是样本的染色体突变位点。通过这种方法确定样本的所有染色体突变位点。
请参阅图3,在本发明的第三实施例中,通过使用并行计算框架spark的分析方法基于高通量测序数据寻找样本的染色体突变位点:首先将包含碱基比对结果的大文件切分为多个小文件块,然后spark s treaming每隔预定时间间隔将从所述文件块中读取到的数据切分为RDD,并将其封装生成job。在多台计算机并行地执行多个job的过程中,通过计算碱基比对质量值(BAQ)过滤掉无效的碱基。最后将多台计算机针对样本上同一位点的并行计算结果进行合并,确定样本的染色体突变位点。
具体地,在本发明的第三实施例中:
第一阈值是指当测序序列碱基的最终质量值小于此值时,认为此碱基无效,并将其过滤掉,不作为寻找样本的染色体突变位点的依据;
第二阈值是指对于样本的某一确定位点,如果突变碱基所占的比率大于等于此值,则确定此位点为样本的染色体突变位点。
Spark:是UC Berkeley AMPLab开发的一种计算框架。
Spark streaming:是建立在Spark上的实时计算框架,通过它提供丰富的API,基于内存的高速执行引擎,用户可以结合流式、批处理和交互式查询应用。
DStream是指离散流(Discretized Stream),它是Spark Streaming提供的基本的抽象,代表一个实时的数据流。
RDD是指弹性分布式数据集(Resilient Distributed Datasets),它是可容错的并行数据结构,使用户能够显式地在内存中保存中间的运算结果,通过控制RDD的分区来优化数据的布局,并使用丰富的转换算子进行操作。
Job是指在spark streaming中,封装了RDD用于进行并行计算的执行实体。
S301、将包含碱基比对结果的大数据文件切分为多个小文件块。
从数据源输入的文件为bam或者sam文件,其中包含了测序序列与参考序列比对结果的大量数据。
多个客户端通过网络上传原始的bam或sam大文件,spark streaming将此大文件根据一定的规则切分成小文件块保存到kafka中。切分规则如下:比如输入的大文件为3G,在spark streaming中将一个block设置为128M,那么输入的大文件总共将被切分为3*1024/128=24块。
S302、参与并行计算的多台计算机并行地将从多个小文件块中读取到的数据切分为RDD,并根据RDD生成并行计算的执行实体job。
参与并行计算的多台计算机上的spark streaming并行地以预定时间间隔(比如3秒)从kafka中保存的多个小文件块中读取数据,并在此过程中将读取到的数据切分为RDD,供spark streaming后续的并行计算使用。参与并行计算的所有计算机按时间片(即预定时间间隔)读取到的所有数据组成了代表持续输入的碱基比对结果的DStream。Spark streaming将对DStream的操作转换为DStream的有向无环图(DAG)。对于每个时间片(即预定时间间隔),spark streaming根据DStream DAG生成RDD DAG,并根据RDD DAG生成相应的执行实体job。后续的步骤S303至S306都是在这里的多个job中进行并行计算的。
S303、确定需要计算碱基比对质量值(BAQ)的碱基。
Spark streaming在参与并行计算的多台计算机上并行地执行步骤S302中生成的多个job。
为了寻找样本的染色体突变位点,需要过滤掉测序序列一部分位点上的碱基:测序数据是基于高通量测序获取的,而高通量测序的错误率在0.01%左右,所以需要去掉低质量的数据(即无效的碱基),不将其作为寻找样本的染色体突变位点的依据。
过滤过程首先需要判断是否需要对碱基计算BAQ值。确定方法如下:如果测序序列与参考序列所有对应位点上的碱基完全一致,则将此测序序列所有位点的碱基作为寻找样本的染色体突变位点的依据,无需计算它们的BAQ值;如果测序序列某一位点与参考序列对应位点上的碱基不一致,则需要对此测序序列所有位点的碱基计算BAQ值。
S304、测序序列与参考序列碱基不一致时计算测序序列所有位点的碱基比对质量值(BAQ)。
请参阅图4,如果测序序列(read序列)和参考序列(reference序列)不完全一致,则对read序列和reference序列建立一个隐马尔可夫模型(HMM),并对read序列中状态为M的碱基计算其碱基比对质量值(BAQ),该值代表此碱基比对结果错误的可能性。
在HMM中,M代表碱基匹配(包括完全一致和错配两种情况),I代表插入,D代表缺失,S代表开始,E代表结束。S指向所有的I和M,所有的M和I指向E。
BAQ算法分为前向递推和后向递推。算法中用到的变量如下:
前向递推:
(a)值的初始化,其中k=1,…,L;i=0,1。
f0,S=1
(b)递推,其中i=2,…,l;k=1,…,L。
对于i=l+1,有:
其中,aij由以下状态转换矩阵确定:
后向递推:
(a)值的初始化,其中k=1,…,L。
bl+1,E=1
(b)递推,其中i=l-1,…,1;k=L,…,1。
当i=1时,δi1=1,其他则为0。
对于i=0,有:
BAQ通过下式得到:
其中,bij由以下状态转换矩阵确定:
S305、确定已计算过BAQ的碱基的最终质量值。
对于已计算过BAQ的碱基,在BAQ和此碱基的测序质量值(测序质量值可自bam或sam文件中获得)中取较小值作为最终质量值。
S306、过滤掉无效的碱基。
在测序序列所有的碱基中,对于已计算过BAQ的碱基,如果其最终质量值小于第一阈值,则认为此碱基无效,并将其过滤掉,不作为寻找样本的染色体突变位点的依据;否则认为此碱基有效,将其保留并作为寻找样本的染色体突变位点的依据。
S307、根据保留下来的碱基计算样本的每个位点上的突变碱基所占的比率。
对于样本上的每一个位点,将所有测序序列在该位点产生的ATCG各碱基的次数(通过对多台计算机上的多个RDD进行并行计算得出)累加,得到该位点上的突变碱基所占的比率。
S308、确定样本的染色体突变位点。
如果样本某一位点上的突变碱基所占的比率大于等于第二阈值,则确定此位点是样本的染色体突变位点;否则确定此位点不是样本的染色体突变位点。通过这种方法确定样本所有的染色体突变位点。
请参阅图5,本发明还提供了一种寻找样本的染色体突变位点的分析装置,所述装置包括:
并行准备模块510,用于根据测序序列和参考序列的比对结果为并行计算准备数据;
并行计算模块520,用于通过并行计算过滤掉准备好的数据中无效的碱基;
结果合并模块530,用于根据过滤后保留的碱基确定样本的染色体突变位点。
所述并行准备模块510包括:
数据切分单元5101,用于对包含所述比对结果的数据进行切分;
执行实体生成单元5102,用于生成包含切分后的数据的执行实体。
优选地,所述数据切分单元5101用于:
将包含所述比对结果的数据的文件切分为文件块;
每隔预定时间间隔将从所述文件块中读取到的数据切分为组成DStream的RDD。
优选地,所述执行实体生成单元5102用于根据RDD生成执行实体job。
所述并行计算模块520包括:
质量计算单元5201,用于:
通过执行实体对准备好的数据进行并行计算:当由样本产生的测序序列与参考序列对应位点上的碱基不一致时,对此测序序列上的每个碱基计算碱基比对质量值,
通过执行实体并行地在所述碱基比对质量值和此碱基的测序质量值中取较小值作为此碱基的最终质量值;
过滤单元5202,用于通过执行实体并行地进行过滤:当所述最终质量值小于此碱基的第一阈值时,过滤掉此碱基。
所述结果合并模块530包括:
比率计算单元5301,用于根据过滤后保留的碱基确定样本的每个碱基位点上的突变碱基所占的比率;
突变位点确定单元5302,用于当所述样本的某一位点上的突变碱基所占的比率大于等于第二阈值时,确定此位点为样本的染色体突变位点。
在本发明的第一实施例中:
并行准备模块510根据碱基的比对结果为并行计算准备数据。并行准备模块510将包含测序序列和参考序列碱基的比对结果的文件切分成多个文件块,保存在文件系统中。并行准备模块510将从文件系统中的多个文件块中读取数据为并行计算进行准备。并行计算模块520通过并行计算过滤掉准备好的数据中无效的碱基。结果合并模块530根据过滤后保留的碱基确定样本的染色体突变位点。
在本发明的第二实施例中:
并行准备模块510中的数据切分单元5101将包含碱基的比对结果的大数据文件切分为多个小数据块。
从数据源输入的文件为bam或者sam文件,其中包含了测序序列与参考序列碱基比对结果的大量数据。
多个客户端通过网络将原始的bam或sam大文件上传到HDFS(Hadoop Distributed File System),并行准备模块510中的数据切分单元5101将此大文件根据一定的规则切分成小数据块并保存。切分规则如下:比如输入的大文件为3G,在spark streaming中将一个block设置为128M,那么输入的大文件总共将被切分为3*1024/128=24块。
并行准备模块510中的执行实体生成单元5102读取HDFS中的小数据块,启动job,job为每一个小文件块生成实际的执行实体map task和reduce task。
并行准备模块510中的执行实体生成单元5102读取HDFS中的小数据块,并启动一个job,job为每一个小数据块生成一个map task和reduce task。Map task和reduce task是并行计算的执行实体。
在map task中,并行计算模块520中的质量计算单元5201通过并行计算碱基比对质量值(BAQ)和最终质量值来过滤掉测序序列位点上无效的碱基,并将过滤后保留下来的碱基保存到HDFS中。
并行计算模块520并行地执行map task和reduce task,先运行map task,然后运行reduce task。
为了寻找样本的染色体突变位点,并行计算模块520中的质量计算单元5201需要并行地计算碱基比对质量值(BAQ)和最终质量值,并行计算模块520中的过滤单元5202根据最终质量值过滤掉测序序列位点上无效的碱基。这一部分并行计算是在map task中进行的。有关计算BAQ和最终质量值以及过滤掉测序序列位点上无效的碱基的算法描述请参见本说明书步骤S303至S306的相关描述。
在reduce task中,结果合并模块530中的比率计算单元5301通过并行计算根据保留下来的碱基计算样本的每一个位点上的突变碱基所占的比率,结果合并模块530中的突变位点确定单元5302根据此比率确定样本的突变位点。
在reduce task中,首先结果合并模块530中的比率计算单元5301从HDFS中读取过滤后保留下来的碱基。然后,对于样本上的每一个位点,比率计算单元5301累加由测序数据产生的ATCG各碱基的次数(通过多台计算机对其上的小数据块进行并行计算得出),从而得到此位点上的突变碱基所占的比率。
结果合并模块530中的突变位点确定单元5302按照以下方法确定样本的染色体突变位点:如果样本某一位点上的突变碱基所占的比率大于等于第二阈值,则确定此位点是样本的染色体突变位点;否则确定此位点不是样本的染色体突变位点。通过这种方法确定样本的所有染色体突变位点。
在本发明的第三实施例中:
并行准备模块510中的数据切分单元5101将包含碱基比对结果的大数据文件切分为多个小文件块。
从数据源输入的文件为bam或者sam文件,其中包含了测序序列与参考序列的碱基比对结果的大量数据。
多个客户端通过网络上传原始的bam或sam大文件,并行准备模块510中的数据切分单元5101将此大文件根据一定的规则切分成小文件块保存到kafka中。切分规则如下:比如输入的大文件为3G,一个block为128M,那么输入的大文件总共将被切分为3*1024/128=24块。
并行准备模块510中的数据切分单元5101将从多个小文件块中读取到的数据切分为RDD,并行准备模块510中的执行实体生成单元5201根据RDD生成并行计算的执行实体job。
并行准备模块510中的数据切分单元5101以预定时间间隔(比如3秒)从kafka中保存的多个小文件块读取数据,并在此过程中将读取到的数据切分为RDD。数据切分单元5101按时间片(即预定时间间隔)读取到的所有数据组成了代表持续输入的碱基比对结果的DStream。数据切分单元5101将对DStream的操作转换为DStream的有向无环图(DAG)。对于每个时间片(即预定时间间隔),数据切分单元5101根据DStream DAG生成RDD DAG。并行准备模块510中的执行实体生成单元510根据RDD DAG生成相应的执行实体job。
并行计算模块520中的质量计算单元5201确定需要计算碱基比对质量值(BAQ)的碱基。
并行计算模块520在参与并行计算的多台计算机上并行地执行前面步骤生成的多个job。
为了寻找样本的染色体突变位点,需要过滤掉测序序列一部分位点上的碱基:测序数据是基于高通量测序获取的,而高通量测序的错误率在0.01%左右,所以需要去掉低质量的数据(即无效的碱基),不将其作为寻找样本的染色体突变位点的依据。
过滤过程首先需要由并行计算模块520中的质量计算单元5201对测序序列的一部分位点的碱基计算碱基比对质量值(BAQ)。质量计算单元5201确定需要计算BAQ的碱基的方法如下:如果测序序列与参考序列所有对应位点上的碱基完全一致,则将此测序序列所有位点的碱基作为寻找样本的染色体突变位点的依据,无需计算它们的BAQ值;如果测序序列某一位点与参考序列对应位点上的碱基不一致,则需要对此测序序列所有位点的碱基计算BAQ值。
当测序序列与参考序列碱基不一致时,并行计算模块520中的质量计算单元5201计算测序序列所有位点的碱基比对质量值(BAQ)。
请参阅图4,如果测序序列(read序列)和参考序列(reference序列)不完全一致,则质量计算单元5201对read序列和reference序列建立一个隐马尔可夫模型(HMM),并对read序列中状态为M的碱基计算其碱基碱基比对质量值(BAQ),该值代表此次碱基比对结果错误的可能性。
在HMM中,M代表碱基匹配(包括完全一致和错配两种情况),I代表插入,D代表缺失,S代表开始,E代表结束。S指向所有的I和M,所有的M和I指向E。
BAQ算法分为前向递推和后向递推。算法中用到的变量如下:
前向递推:
(a)值的初始化,其中k=1,…,L;i=0,1。
f0,S=1
(b)递推,其中i=2,…,l;k=1,…,L。
对于i=l+1,有:
其中,aij由以下状态转换矩阵确定:
后向递推:
(a)值的初始化,其中k=1,…,L
bl+1,E=1
(b)递推,其中i=l-1,…,1;k=L,…,1
当i=1时,δi1=1,其他则为0。
对于i=0,有:
BAQ通过下式计算得到:
其中,bij由以下状态转换矩阵确定:
并行计算模块520中的质量计算单元5201确定已计算过BAQ的测序序列所有碱基的最终质量值。
对于已计算过BAQ的测序序列的碱基,质量计算单元5201在BAQ和此碱基的测序质量值(测序质量值可自bam或sam文件中获得)中取较小值作为最终质量值。
并行计算模块520中的过滤单元5202过滤掉无效的碱基。
并行计算模块520中的过滤单元5202按照以下方法过滤掉测序序列上无效的碱基:在测序序列所有的碱基中,对于已计算过BAQ的碱基,如果其最终质量值小于第一阈值,则认为此碱基无效,并将其过滤掉,不作为寻找样本的染色体突变位点的依据;否则认为此碱基有效,将其保留并作为寻找样本的染色体突变位点的依据。
结果合并模块530中的比率计算单元5301根据保留下来的碱基计算样本的每个位点上的突变碱基所占的比率。
对于样本上的每一个位点,结果合并模块530中的比率计算单元5301累加所有测序序列在该位点产生的ATCG各碱基的次数(通过对多台计算机上的多个RDD进行并行计算得出),从而得到此位点上的突变碱基所占的比率。
结果合并模块530中的突变位点确定单元5301按照以下方法确定每一个位点是否为突变位点:如果样本某一位点上的突变碱基所占的比率大于等于第二阈值,则确定此位点是样本的染色体突变位点;否则确定此位点不是样本的染色体突变位点。突变位点确定单元5301通过这种方法确定样本所有的染色体突变位点。
本领域普通技术人员可以理解,实现上述本发明实施例中的基于高通量测序数据寻找样本的染色体突变位点的分析方法和分析装置可以通过程序指令相关的硬件来完成,所述的程序可以存储于可读取存储介质中,该程序在执行时执行上述方法中的对应步骤。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原来的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!