基于全部连续列一致演化型的时序基因芯片数据挖掘方法与流程
本发明属于计算机算法,数据挖掘技术领域,具体涉及一种针对时序基因芯片表达数据的基于全部连续列一致演化型的时序基因芯片数据挖掘方法。
背景技术:
在生物信息领域,科技的飞速发展,生命科学的不断进步,使得利用数据挖掘技术来分析生物信息已经成为了当前和未来的趋势。生物信息学分析的要点是基因组学及蛋白组学两个领域,分别把核酸序列及蛋白质序列当做出发点,把高通量分析当做技术点,研究序列蕴含的生物意义(campbell&heyer,2003)。人类基因组计划的开启促进了生物信息学的前进,随着基因组测序工作的结束,后基因组时代的大门开启了(masters&lakhani,2000)。
有一种基因表达数据矩阵叫做时序基因表达数据矩阵,这种数据是时序数据,故数据中有时间的因素。时序基因表达数据矩阵的值是测试基因在不一样的时间下的表达而得到的,矩阵中的值揭示了表达值随着时间的不同而不同的特点(bar-joseph,2004)。利用基因测试技术去获得连续时间的基因表达程度,能够学习到基因和时间变化的变化关系,从而可以学习到不同基因的各种联系,基因之间的调控联系等等的信息(korenberg,2007)。对于共调控的基因,它们可能在某个连续时间段内有一样的表达水平,此连续时间段可能和某个细胞过程的一个阶段密切相关(zhang,zha,&chu,2005)。时序基因表达数据往往能够提供一些生理进程的共同调控的知识,并且能够反映生物过程和时间变化的关系,故时序基因表达数据对分析基因调控的网络与动态的生物过程有重要作用(bar-joseph,2004;f.liu&wang,2010)。
在时序基因表达数据矩阵中,行表示基因,列表示先后的实验时间,有时间的先后顺序。对于时间序列数据的时序基因表达数据,能够发现相邻时间的基因表达水平的规律会更有意义。但是很多双聚类模型挖掘到的双聚类不是连续相邻时间,故这些双聚类模型并不能很好应对时序数据。随着研究的深入,有一些双聚类方法能够针对时序数据的特点来挖掘,这些方法能够分析时序基因表达数据矩阵,发现连续时间设置的生物知识。
聚类是数据挖掘领域的经典方法,当今研究基因表达数据的一个重要方案是使用聚类方法,例如k-均值(j.a.hartigan&wong,1979),层次聚类(cameron,middleton,chenn,&olson,2012),自组织神经网络(
分析基因表达数据应用传统聚类还有部分不足之处。第一,聚类方法是考虑一个维度的全部值,或者只是考虑行而形成行的聚类,或者只是考虑列而形成列的聚类,形成的聚类考虑的都是全局的信息。但是对于细胞过程研究,可能一些基因只是在一些环境设置下有明显的反应,而不是在全部的环境设置都有反应。第二,传统聚类一般把样本划分成没有交集的集合,及一个样本或基因只能最多属于一个聚类,而不能同时属于多个聚类(tanay,sharan,&shamir,2002)。但是实际基因研究中,研究的基因有很大机会参与若干生物过程,即该基因很大机会同时是若干不同聚类的一员。
因为传统聚类的局限,使得传统聚类难以挖掘数据矩阵中的局部表达模式,难以挖掘到基因之间的隐藏的复杂关系(cheung,cheung,kao,yip,&ng,2006),而双聚类可以解决这个问题。
为了克服聚类的缺点,双聚类应运而生并被广泛的基因数据研究所采纳(eren,deveci,
cheng和church(cheng&church,2000)第一次把双聚类的概念使用到基因表达数据的研究中来,并提出了用来限定双聚类的均方残差,同时提出启发式方法cc方法用来挖掘双聚类。cc方法主要过程是计算子矩阵的均方残差,不断对行和列进行删除,最后挖掘到均方残差较小的双聚类。他们的方法能够找到基因表达数据的一些生物信息,但是难以分析时序基因表达数据,因为时序基因表达数据有内部的时间信息,而该方法不能发现时间序列的联系。
zhang等人(zhangetal.,2005)改进cc方法而提出可以用来分析时间序列数据的方法cc-tsb。cc-tsb双聚类方法的主要思想和cc方法类似,主要区别是cc-tsb方法对列的操作有限制,保证双聚类的列是连续若干列,添加或去除列是在首列或尾列操作,所以该方法能够发现一定的时间信息的双聚类。但是,cc-tsb双聚类方法使用的限制子矩阵的量也是cheng和church提出的均方残差,但是均方残差关注的是基因表达值的绝对大小,而噪声因素,量纲因素都影响基因表达值,故cc-tsb方法的对噪声数据的鲁棒性不够强。另外,均方残差函数没有关注内部序列关系,故这种方法并不能很好衡量时间序列基因表达数据的特点。
考虑连续时间的因素,关注“相邻时间之间的一致变化(coherentevolution)趋势”比关注“实际值的大小”更有意义(saracmadeira&arlindololiveira,2004),一种连续一致列(contiguouscoherentcolumns,简称ccc)被sarac.madeira等人(madeira&oliveira,2005)提出。ccc将模式限制在连续的列上,用于寻找所有最大连续列一致双聚类(contiguouscolumncoherentbicluster)。ccc方法首先把原始数据转化为代表升降的字符型序列,然后利用后缀树数据结构来提高效率,最后挖掘到最大连续列一致双聚类。
因为ccc方法并没有考虑噪声的影响,后来考虑噪声因素,sarac.madeira等人改进原来的ccc双聚类方法,提出e-ccc双聚类方法(madeira&oliveira,2007)。e-ccc双聚类方法和ccc双聚类方法类似,不同之处是e-ccc方法能够容忍一定的误差的情形,把误差小于预定阈值的模式看作同一个模式。实验结果说明e-ccc方法对噪声的鲁棒性更加好,找到的双聚类的生物信息更加富集。
ccc、e-ccc方法都考虑到了时序基因数据的重要特点——时序性,关注相邻两个时间点之间的变化趋势,并且关注基因表达值的相对大小而非绝对大小,故模型有较强的噪声鲁棒性。但是,ccc只考虑局部最长的模式,而丢失了第二长,第三长等等其他长度的连续列一致演化型的信息。另外,现有的连续列一致演化型的寻找方法ccc、e-ccc都是基于后缀树的字符串处理技术,空间复杂度比较高,难以处理数据规模庞大的数据。
现有方法存在如下不足:
(1)现有的方法没有考虑把相邻时间的信息考虑进来,但是时序数据包含先后时间顺序,在连续时间有先后序列关系,不能很好衡量该类数据的相似性。因此,需要解决考虑相邻时间信息的双聚类挖掘问题。
(2)现有方法只考虑局部最长的模式,而丢失了第二长,第三长等等其他长度的连续列一致演化型的信息。因此,需要解决时序基因表达数据两个序列间全部连续列一致演化型的信息的捕捉问题。
(3)现有的方法的操作效率较低,在挖掘双聚类的过程中需要耗费较多的时间和资源,这也是本发明所要解决的技术问题之一。
技术实现要素:
本发明的目的在于克服现有技术存在的上述不足,提出一种基于连续列一致演化性的基因芯片表达数据的双聚类挖掘方法。首先将原始矩阵转化为一个差分矩阵,然后提出新的衡量双聚类的质量的方法,接着通过改变行列来迭代得到双聚类。该方法考虑了相邻时间的因素,而且能够捕捉到全部的连续列一致演化型的信息,并比现有方法高效快速,具体技术方案如下。
基于全部连续列一致演化型的时序基因芯片数据挖掘方法,其包括如下步骤:
(1)输入时序基因芯片数据,对数据进行预处理;
(2)使用基于全部连续列一致演化型的衡量双聚类方法,对步骤(1)得到的数据,进行初始化双聚类;
(3)使用基于全部连续列一致演化型的衡量双聚类方法,通过增删行列的方式,迭代更新双聚类;
(4)利用行列阈值限制输出,得到双聚类,实现时序基因芯片数据挖掘。
进一步地,步骤(1)具体是:数据初始化阶段,首先将原始矩阵转化为一个记录任意两个相邻时间变化趋势的矩阵,用于反映每一个基因的表达值在相邻两个时间点下的变化趋势。
进一步地,步骤(2)(3)中,基于全部连续列一致演化型的衡量双聚类方法的具体操作是:计算双聚类中每行和双聚类核心的全部连续列一致演化型数量,然后对这些数量求和,接着用求和结果除以双聚类的列数,从而得到衡量双聚类的值,记为peraccc。
进一步地,步骤(2)(3)中,所述计算双聚类中每行和双聚类核心之间的全部连续列一致演化型数量的具体操作是:在所要计算的两行中,将任一连续列中两行表达值具有相同变化趋势的模式作为统计对象,统计所有所述模式的个数,得到的结果记为accc。
进一步地,步骤(2)具体是:第一步:选择双聚类核心,即在数据矩阵中随机选择一行,然后随机选择一个连续列作为双聚类的列集,所述行在该连续列上的值共同构成双聚类核心;第二步:得到双聚类的行集,即在数据矩阵中随机选择若干行作为初始行集,对所述初始行集的每一行分别和第一步得到的双聚类核心计算所述accc,把大于设定值的accc所对应的行保存到双聚类的行集;第三步:更新双聚类核心,即对双聚类的每一列,计算每一列在所述双聚类的行集上的众数,用所述众数作为双聚类核心在对应列的值,从而得到更新后的双聚类核心。
进一步地,步骤(3)具体是:第一步:计算得到给定双聚类的peraccc值;第二步:使用添加和删除的方式对列进行更新;第三步:通过扫描数据矩阵添加满足条件的行对行进行更新;第四步:通过与步骤(2)中第三步相同的取众数的方法更新双聚类核心;第五步:根据更新后的行集、连续列、双聚类核心计算更新后双聚类的peraccc值;第六步:计算更新双聚类前后的双聚类的peraccc值的变化率,判断是否满足预设阈值,以确定是否继续迭代更新双聚类。
进一步地,步骤(3)中,使用添加和删除的方式对列进行更新,连续列的添加条件和删除条件如下:
列添加条件:如果添加一列后,计算所得的peraccc值增加,则确认添加此列;
首先向右拓展添加连续列,拓展添加结束后,继续向左拓展添加连续列,直到拓展添加结束;
列删除条件:如果删除一列后,计算所得的peraccc值增加,则确认删除此列;
首先从最左边的列开始向右进行删除操作,直到停止;然后从最右边开始向左进行删除操作,直到停止;
如果一个连续列向右拓展添加了若干列,那么不用删除右边的列;如果一个连续列向左拓展添加了若干列,那么不用删除左边的列。
进一步地,步骤(3)中,通过扫描数据矩阵添加满足条件的行对行进行更新,行添加条件如下:
行更新条件:如果添加一行后,计算所得的peraccc值大于或等于没有添加该行之前双聚类的peraccc值,则确认添加此行,否则不添加;
首先对双聚类计算得到peraccc值,然后扫描一遍数据矩阵,分别计算每行和双聚类核心之间的accc值,然后把所述accc值除以双聚类核心的连续列的列数得到值,添加所述值大于或等于所述peraccc值对应的行到行集中,从而更新行集。
进一步地,步骤(3)中所述是否继续迭代的判断方法具体是:设定一个变化率阈值,把没有更新行列和核心之前计算得到的peraccc值记为pa0,把更新之后的计算得到的peraccc值记为pa1,如果pa0到pa1的变化率小于预设的变化率阈值,则停止迭代更新双聚类,进入步骤(4)。
进一步地,步骤(4)具体是:设定一组行列阈值,如果双聚类不满足预设的行列阈值,则重新初始化双聚类,转到步骤(2);如果双聚类满足行列阈值,则得到双聚类结果,实现时序基因芯片数据挖掘。
本发明与现有技术相比,具有如下突出实质性特点和显著优点:
本发明中,对时序基因芯片表达数据使用连续列一致演化型模型进行分析,找到满足本发明提出的衡量双聚类的方法的子矩阵。本发明考虑数据中有时间的因素,能够学习到基因和时间变化的变化关系,从而可以学习到不同基因的各种联系,基因之间的调控联系等等的信息。对于共调控的基因,它们在某个连续时间段内有一样的表达水平,此连续时间段和某个细胞过程的一个阶段密切相关,能够提供一些生理进程的共同调控的知识,对分析基因调控的网络与动态的生物过程有重要作用。另外一方面,本发明的操作效率很高,能在使用较少资源且在较短的时间内完成双聚类的挖掘。
附图说明
图1是基于全部连续列一致演化型的基因芯片数据挖掘方法的流程图。
图2是初始化双聚类例子的流程图。
图3是更新双聚类列集例子的流程图。
图4是更新双聚类行集例子的流程图。
图5是迭代更新及判断例子的流程图。
图6是挖掘到双聚类的折线图。
图7是酵母菌芯片数据富集性分析的柱状图。
图8是操作时间与数据矩阵行数关系的折线图。
图9是操作时间与数据矩阵列数关系的折线图。
具体实施方式
下面结合附图对本发明的具体实施方式作进一步说明,但本发明的实施不限于此。需特别指出的是,以下若有未特别详细说明的符号或操作过程,均是本领域技术人员可以参照现有技术实现的。
如图1,本实例的基于连续列一致演化性的基因芯片表达数据的双聚类挖掘方法包括如下内容:
1、输入时序基因芯片数据,对数据进行预处理。
数据初始化阶段,首先将原始矩阵转化为一个记录任意两个相邻时间变化趋势的矩阵,用于反映每一个基因的表达值在相邻两个时间点下的变化趋势。
在这个例子中,将变化趋势简化为两种情况,一种为增加或不变,另一种为减少,分别用1,0表示,最终把原始矩阵转化为列数比原始矩阵少1列的矩阵,称为差分矩阵。
原始矩阵如表1所示,在表1中,行标a、b、c、d表示不同的基因,列标t1、t2、t3、t4、t5、t6表示时间点。表1经过预处理得到的差分矩阵如表2所示,表2中,行标a、b、c、d表示不同的基因,列标1、2、3、4、5分别表示t1到t2、t2到t3、t3到t4、t4到t5、t5到t6的变化趋势。例如表1行a,t1,t2时刻表达值分别是0.55和0.19,因为0.19<0.55,所以表2矩阵中行a的第一列值为0。同理,表1中行a中0.83>0.19,所以行a第二列值为1。
表1.转换前的原始时间序列数据
表2.转换后的差分矩阵数据
2、使用基于全部连续列一致演化型的衡量双聚类方法,对步骤(1)得到的数据,进行初始化双聚类。
基于全部连续列一致演化型的衡量双聚类方法的具体操作是:计算双聚类中每行和双聚类核心的全部连续列一致演化型数量,然后对这些数量求和,接着用求和结果除以双聚类的列数,从而得到衡量双聚类的值,记为peraccc。
所述计算双聚类中每行和双聚类核心之间的全部连续列一致演化型数量的具体操作是:在所要计算的两行中,将任一连续列中两行表达值具有相同变化趋势的模式作为统计对象,统计所有所述模式的个数,得到的结果记为accc。
在将变化趋势简化为两种情况的例子中,计算两行的全部连续列一致演化型个数的方法具体操作如下:
首先生成和所述两行相同长度一行数据,记为记录行。然后对所述两行,依次观察每列,两行的值都相同则记录行在该列的值置为1,不同则置为0,从而更新记录行。对记录行,统计全1出现的段数,和每段中1出现的个数,最后通过计算出连续列一致演化型个数,从而得到结果。全1出现的段指的是值全部都是1的部分。
例如:记录行是:110011101。全1出现的段共有3段。每一段用下划线表示,分别是:110011101,故每一段1的个数分别是:2,3,1,通过计算得到的相似度是:10。
初始化双聚类具体是:第一步:选择双聚类核心,即在数据矩阵中随机选择一行,然后随机选择一个连续列作为双聚类的列集,所述行在该连续列上的值共同构成双聚类核心;第二步:得到双聚类的行集,即在数据矩阵中随机选择若干行作为初始行集,对所述初始行集的每一行分别和第一步得到的双聚类核心计算所述accc,把大于设定值的accc所对应的行保存到双聚类的行集;第三步:更新双聚类核心,即对双聚类的每一列,计算每一列在所述双聚类的行集上的众数,用所述众数作为双聚类核心在对应列的值,从而得到更新后的双聚类核心。
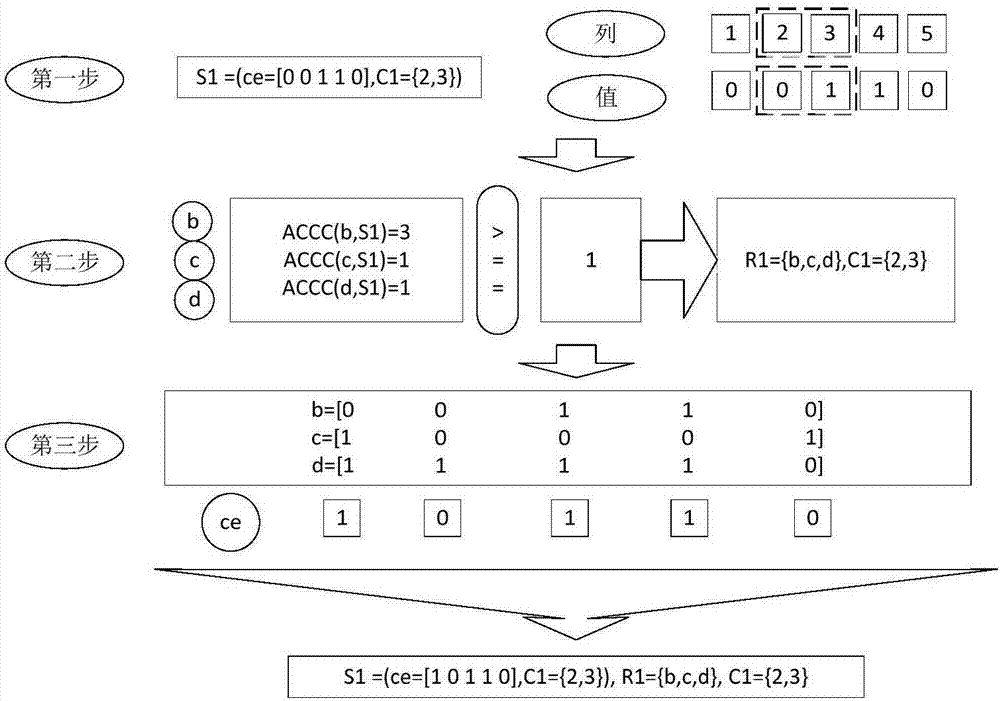
初始化双聚类例子如图2所示。
第一步:选择双聚类核心,即在数据矩阵中随机选择一行,然后随机选择一个连续列作为双聚类的列集,所述行在该连续列上的值共同构成双聚类核心。在表2的差分矩阵中随机选择的一行是行b,即[00110],随机选择的连续列是2,3列,构成双聚类核心。如图2所示,s1表示双聚类核心,ce表示双聚类核心的值,即行b的值,c1表示双聚类核心的连续列,为2,3列。图2中,“列”对应的那行表示差分矩阵的列标,“值”对应的那行表示双聚类核心的值,虚线框内的列表示选择的连续列及对应值。
第二步:得到双聚类的行集,即在数据矩阵中随机选择若干行作为初始行集,对所述初始行集的每一行分别和第一步得到的双聚类核心计算所述accc,把大于或等于设定值的accc所对应的行保存到双聚类的行集。随机选择的初始行集是{b,c,d},对初始行集{b,c,d}的每一行分别和第一步得到的双聚类核心计算accc,结果分别是3,1,1,设定的阈值1,因为3,1,1都大于或等于1,故得到双聚类的行集r1={b,c,d}。
第三步:更新双聚类核心,即对双聚类的每一列,计算每一列在所述双聚类的行集上的众数,用所述众数作为双聚类核心在对应列的值,从而得到更新后的双聚类核心。如图2所示,图中显示了行b,c,d的值,对每列取众数得到ce对应的行,值为[10110]。例如行b,c,d的第一列,0的个数是1,而1的个数是2,即1的个数大于0的个数,众数是1,所以核心的第一列为1。
经过上述步骤操作之后,初始的双聚类的行集列集及双聚类核心分别是:s1=(ce=[10110],c1={2,3}),r1={b,c,d},c1={2,3}。
3、使用基于全部连续列一致演化型的衡量双聚类方法,通过增删行列的方式,迭代更新双聚类。
第一步:计算得到给定双聚类的peraccc值;第二步:使用添加和删除的方式对列进行更新;第三步:通过扫描数据矩阵添加满足条件的行对行进行更新;第四步:通过与步骤(2)中第三步相同的取众数的方法更新双聚类核心;第五步:根据更新后的行集、连续列、双聚类核心计算更新后双聚类的peraccc值;第六步:计算更新双聚类前后的双聚类的peraccc值的变化率,判断是否满足预设阈值,以确定是否继续迭代更新双聚类。
第一步:计算得到给定双聚类的peraccc值。
操作过程如基于全部连续列一致演化型的衡量双聚类方法的具体操作所述。
第二步:使用添加和删除的方式对列进行更新。
使用添加和删除的方式对列进行更新,连续列的添加条件和删除条件如下:
列添加条件:如果添加一列后,计算所得的peraccc值增加,则确认添加此列;
首先向右拓展添加连续列,拓展添加结束后,继续向左拓展添加连续列,直到拓展添加结束;
列删除条件:如果删除一列后,计算所得的peraccc值增加,则确认删除此列;
首先从最左边的列开始向右进行删除操作,直到停止;然后从最右边开始向左进行删除操作,直到停止;
如果一个连续列向右拓展添加了若干列,那么不用删除右边的列;如果一个连续列向左拓展添加了若干列,那么不用删除左边的列。
更新双聚类的列集过程的例子如图3所示。
双聚类的行集r1={b,c,d},连续列c1={2,3},首先计算该双聚类的peraccc值,记pa=peraccc(r1,c1),计算得pa=0.83,如图中“更新前”所示。向右拓展添加连续列。首先考察添加第四列,如图3中步骤1所示,下划线的列是当前考察添加的列,这一步为第4列,即连续列为{2,3,4},计算peraccc值为1.11,因为1.11>pa=0.83,所以确认添加第4列到连续列c1,把新的peraccc值1.11赋值给pa。继续向右拓展连续列,添加第5列,如图中步骤2所示,和考察第4列时候的方法相同,即连续列为{2,3,4,5},计算peraccc值为1.42,因为1.42>pa=1.11,所以确认添加第5列到连续列c1,把新的peraccc值1.42赋值给pa。因为第5列是最右边的列,不能再向右拓展了,所以停止向右拓展。向右拓展停止后,开始向左拓展。首先考察添加第1列,如图中步骤3所示,即连续列为{1,2,3,4,5},计算peraccc值为1.33,因为1.33<pa=1.42,所以不添加第4列到连续列c1,同时pa保持原值1.42。因为第1列是最左边的列,不能再向左拓展了,所以停止向左拓展。
添加步骤结束后,开始连续列删除步骤。首先考虑删除最连续列中最左边的列第2列,如图中步骤4所示,删除线的列是当前考虑删除的列,即连续列为{3,4,5},计算peraccc值为1.44,因为1.44>pa=1.42,所以确认从连续列c1删除第2列,把新的peraccc值1.44赋值给pa。继续考察删除第3列,如图中步骤5所示,即连续列为{4,5},计算peraccc值为1,因为1<pa=1.44,所以不删除第4列,同时pa保持原值1.44。因为初始化后的连续列为{2,3},经过列拓展添加后的连续列是{2,3,4,5},即在向右拓展添加过程中确认添加了4,5列到连续列,所以不必进行从右往左的删除列的操作。所以这一轮操作最后生成的连续列c1={3,4,5},如图中“更新后”所示。
第三步:通过扫描数据矩阵添加满足条件的行对行进行更新.
通过扫描数据矩阵添加满足条件的行对行进行更新,行添加条件如下:
行更新条件:如果添加一行后,计算所得的peraccc值大于或等于没有添加该行之前双聚类的peraccc值,则确认添加此行,否则不添加;
首先对双聚类计算得到peraccc值,然后扫描一遍数据矩阵,分别计算每行和双聚类核心之间的accc值,然后把所述accc值除以双聚类核心的连续列的列数得到值,添加所述值大于或等于所述peraccc值对应的行到行集中,从而更新行集。
更新双聚类行集的过程的例子如图4所示。
在以上步骤中得到的支持行集为r1={b,c,d},连续列为c1={3,4,5},双聚类核心是s1=(ce=[10110],c1={3,4,5}),把经过更新列之后双聚类的peraccc值赋给pa0,即pa0=1.44.扫描差分矩阵中所有的行,首先考察第1行行a,peraccc({a},c1)=1,因为1<pa0=1.44,所以不添加行a到行集r1.接着考察第2行行b,peraccc({b},c1)=2,因为2>pa0=1.44,所以添加行a到行集r1.同理,考察行c,d。最后得到行集r1={b,d}。相比于原双聚类的行集{b,c,d},相当于行c不满足条件,从而行c被删除了。
第四步:通过与步骤(2)中第三步相同的取众数的方法更新双聚类核心.
更新双聚类的方法和上文中提到的一样,因为更新双聚类核心的方法已经在上文中详细叙述了,所以这里不展开叙述。更新双聚类,得到的核心是s1=(ce=[10110],c1={3,4,5})。
第五步:根据更新后的行集、连续列、双聚类核心计算更新后双聚类的peraccc值。
因为计算peraccc的方法已经在上文中详细叙述了,所以这里不展开叙述。
第六步:计算更新双聚类前后的双聚类的peraccc值的变化率,判断是否满足预设阈值,以确定是否继续迭代更新双聚类。
是否继续迭代的判断方法具体是:设定一个变化率阈值,把没有更新行列和核心之前计算得到的peraccc值记为pa0,把更新之后的计算得到的peraccc值记为pa1,如果pa0到pa1的变化率小于预设的变化率阈值,则停止迭代更新双聚类,进入步骤(4)。
迭代更新及判断的例子如图5所示。
在这个例子中,不妨设变化率阈值为deta=0.1,即peraccc的值的变化率需要小于0.1才停止迭代.如图5所示。没有更新双聚类之前双聚类的peraccc值pa0=0.83,更新双聚类后,计算得到双聚类的peraccc值pa1=2。计算(pa1-pa0)/pa0=1.41,因为1.41>deta=0.1.第二次迭代更新的操作和第一次的操作一样,这里不再展开叙述.第二次迭代中,没有更新双聚类之前双聚类的peraccc值pa0=2,更新双聚类后,计算得到双聚类的peraccc值pa1=2.计算(pa1-pa0)/pa0=0,因为0<deta=0.1,所以停止迭代,得到双聚类的行集列集(r1,c1)=({b,d},{3,4,5}).
4、利用行列阈值限制输出,得到双聚类,实现时序基因芯片数据挖掘。
设定一组行列阈值,如果双聚类不满足预设的行列阈值,则重新初始化双聚类,转到步骤(2);如果双聚类满足行列阈值,则得到双聚类结果,实现时序基因芯片数据挖掘。
在这个例子中,不妨设行阈值min_row=2,列阈值min_col=2。双聚类的行集列集分别是(r1,c1)=({b,d},{3,4,5})。显然双聚类行数为3>min_row=2,故输出。故最终的双聚类结果是(r1,c1)=({b,d},{3,4,5})。输出的双聚类的折线图如图6所示。
参照上述流程,以下再举一个例子。
1、数据矩阵来源
数据矩阵是酵母菌芯片数据,该数据是时序基因芯片数据,来自cho等人(r.j.choetal.,1998)的酵母菌测试数据。采样间隔为10分钟,共有17个采样点,记录了基因的表达水平。该数据矩阵的行代表基因在不同时间点的表达,列代表同一时间点下所有基因的表达情况。对于该数据矩阵中的缺失值,用“三次样条插值方法”进行填补,得到的数据矩阵的大小为6147×17。
人工合成数据矩阵是随机合成的数据,数据的取值范围是0到1,服从均匀分布。生成的若干数据集,其中一组数据集是列数固定为20列,行数从1000行增加到2000行,增幅为100行,另外一组数据集是行数固定为1000,列数从20列增加到40列,增幅是2列。
2、对挖掘到的双聚类结果进行go分析
go(geneontology)注释被用来检验双聚类的生物学意义,对产生的聚类结果作真实性的验证。用gotoolbox对得到的双聚类结果进行生物意义分析,利用了p-value。通过计算已找到的模式与已知基因类别之间的p-value,观察属于同一类基因的geneontology注释的相似性,可以知道双聚类的生物意义。基因表达数据相似的基因通常属于同一生物学通路,并且生物过程和分子功能具有相似之处。对找到的双聚类进行go(geneontology)注释实验,得到的p-value值越小,说明已找到的模式与已知基因类别的关联性越强。
酵母菌芯片数据的go分析部分结果如表3所示,表3中的第一行是go:0002181,基因数据库中总共7166个基因,其中仅186个基因(占2.6%)属于细胞质翻译的go。基于全部连续列一致演化型的时序基因芯片数据挖掘方法找到的一个双聚类有754个基因,其中有90个基因(占11.9%)属于细胞质翻译的go项。计算得到非常小的p-value值(4.62e-37),这个非常小的p-value值反映了这个双聚类的显示显著的生物意义。在酵母菌芯片数据中,go分析的结果显示了找到的双聚类结果具有显著的生物意义。
表3.双聚类结果的go分析部分结果表
3、对挖掘到的双聚类进行生物富集性分析
为了从统计意义上验证得到的双聚类的生物意义,通过分析双聚类结果的生物功能富集性来进行生物意义分析(al-akwaa&kadah,2009;
在酵母菌芯片数据上做生物富集性分析,对比多种聚类或双聚类方法,双聚类结果的富集比柱状图如图7所示,由图可知基于全部连续列一致演化型的方法得出的生物富集性要明显高于ccc、cc-tsb、ccc,opsm、xmotifs,k-means这几个方法。由真实基因表达数据集的富集性分析所示,基于全部连续列一致演化型的时序基因芯片数据挖掘方法很好地从基因表达数据获得更有价值的具有生物学意义的双聚类。
4、对挖掘方法进行可扩展性分析
分析了程序运行时间和数据集大小的关系.图8和图9展示了方法在不同的数据矩阵的尺寸下的运行时间情况,从图中可以看到在图8和图9表现了相似的趋势。对于图8,数据矩阵的总列数为固定的20。方法程序中,矩阵的行数从1000增加到2000,每次增加100,从图8可以看到,随着行数的增加,方法的运行时间线性增大。对于图9,数据矩阵的总行数为固定的2000。方法程序中,矩阵的列数从20增加到40,每次增加2列。从图9可以看到,随着列数的增加,方法的运行时间近似线性增大。
因此,从以上的实验结果可以看到,基于全部连续列一致演化型的时序基因芯片数据挖掘方法在数据量不断增加的情况具有良好的扩展性能。
上述实例进一步说明本发明具有如下优点:
(1)该方法把相邻时间的信息考虑进来,能够有效挖掘到具有时间序列信息的双聚类。能够学习到基因和时间变化的变化关系,可以学习到不同基因的各种联系,基因之间的调控联系等等的信息。
(2)该方法考虑了全部的连续列一致演化型的信息,不会丢失较短长度的连续列一致演化型的信息。能够有效找到具有显著生物意义的双聚类,提供一些生理进程的共同调控的知识。
(3)该方法的操作效率很高,能在使用较少资源且在较短的时间内完成双聚类的挖掘,方法的操作时间和数据规模近似成线性关系,不会随着数据规律增大而急剧增长,具有现实的实践意义。
- 还没有人留言评论。精彩留言会获得点赞!