一种基于多特征融合的药物副作用预测方法与流程
本发明涉及生物信息学,尤其是一种基于多特征融合的药物副作用预测方法,该方法基于药物的四种特性,使用多标签集成k近邻方法预测药物副作用。
背景技术:
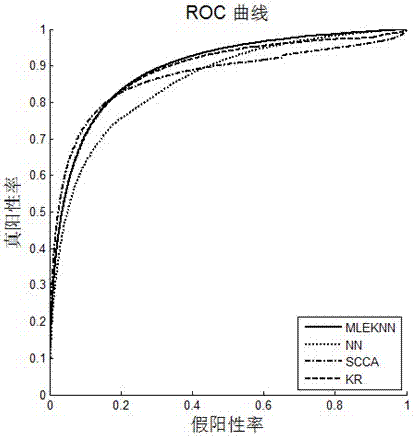
:近年来,药物领域的安全性问题引起了国内外的广泛重视。研究人员开始借助模式识别、复杂网络理论等手段从海量的药物相关数据信息中挖掘新的潜在药物副作用信息和评估药物的安全性。迄今为止,国内外研究人员大致提出了四类药物副作用预测方法:基于统计的方法、基于分子对接的方法、基于文本挖掘的方法和基于机器学习的方法。fukuzaki等人在2009年利用统计相似性模型基于药物的通路(pathway)和基因表达谱的信息来预测药物副作用(参见文献fukuzakim,sekim,kashimah,etal.sideeffectpredictionusingcooperativepathways[c]//ieeeintconfbioinformaticsbiomed,2009:142-147)。但是这一方法要求检测到药物发生化学扰动的情况下的基因表达情况,较难实现。同时通路信息复杂多样,仅根据基因表达谱数据对通路进行统计学分类,容易丢失药物化合物结构等很多重要信息。随后lounkine等人在2012年使用大规模统计模型基于药物的化学结构来预测一个化合物是否会与偏离靶标相绑定。但是他们仅依赖相似性集成方法,去寻找化合物和那些配体之间的相似性,来预测偏离靶标的活动(参见文献lounkinee,keisermj,whitebreads,mikhailovd,hamonj,jenkinsjl,etal.large-scalepredictionandtestingofdrugactivityonside-effecttargets.nature,(2012)486:361-367)。liu等人在2014年基于药物化学结构学习法对现有药物的不良反应进行了因果关系分析(参考文献lium,cair,huy,etal.determiningmolecularpredictorsofadversedrugreactionswithcausalityanalysisbasedonstructurelearning.jammedinformassoc.,(2014),21(2):245-251.),但并没有进一步扩展应用到新药物的预测中。使用统计学分析的方法预测药物副作用,优点是样本信息量大,比较有说服力。缺点是预测的结果往往受统计方法的影响很大。simon等人通过计算药物与偏离靶标蛋白结合口袋的对接分值(参考文献simonz,peragovicsvigh-smellerm,etal.drugeffectpredictionbypolypharmacology-basedinteractionprofiling.jcheminfmodel.,(2011),52(1):134-145.),与药物最初目标靶标的最高对接分值之间的相似性来预测副作用。基于分子对接的药物副作用预测优点是直接使用dock等软件模拟计算药物和靶标蛋白之间结合的概率分值,模拟副作用产生机制,预测较直接。但是药物靶标结合口袋的对接分值计算花费时间较长,效率往往较低。patki等人分别基于文本挖掘的方法预测药物的副作用(参考文献patkia,sarkera,pimpalkhutep,etal.miningadversedrugreactionsignalsfromsocialmedia:goingbeyondextraction.proceedingsofbiolinksig,2014,1-8.)。他们首先对于药物有关的文献进行分类和标记,然后通过实体识别和关系提取等自然语言处理方法来分析和挖掘药物可能存在的副作用。然而基于文本挖掘的方法缺点是,前期准备需要大量的人力,且与药物有关的大量临床文献并不容易获得。pauwel等人(参考文献pauwelse,stovenv,yamanishiy.predictingdrugside-effectprofiles:achemicalfragment-basedapproach.bmcbioinformatics,2011,12(1):169.),利用典型相关分析法,分别基于药物的化学结构及靶标蛋白来预测副作用,典型相关分析法可以结合高维异构特征,在药物的化学结构特征和副作用特征之间寻找相关组件进而预测副作用。然而典型相关分析更适合预测与特殊药物特性相关的特殊种类的副作用,并不能准确预测类似头痛等发生频率较高但真实存在的副作用。随后yaminishi等人在2012年使用核回归方法整合了药物的化学结构和药物的靶标蛋白信息来预测副作用(参考文献yamanishiy,pauwelse,koteram.drugside-effectpredictionbasedontheintegrationofchemicalandbiologicalspaces.jcheminfmodel.,2012,52(12):3284-3292.)。然而其整合只是将不同的药物特征简单的叠加,没有进一步进行特征的筛选与优化。iwata等人在2013年使用逻辑回归方法首次阐述了药物的副作用与药物的靶标蛋白的蛋白质结构域之间可能存在的联系(参考文献iwatah,mizutanis,tabeiy,etal.inferringproteindomainsassociatedwithdrugsideeffectsbasedondrug-targetinteractionnetwork[j].bmcsystbiol.,2013,7:s6-18.)。然而其并没有进一步使用结构域信息进行新药的副作用预测。zhang等人2015年利用集成学习的方法融合了包括药物的化学特性、生物特性和表型特性在内的多个药物特征来预测副作用(参考文献zhangw,liuf,luol,etal.predictingdrugsideeffectsbymulti-labellearningandensemblelearning[j].bmcbioinformatics,2015,16(1):365.)。但是其特征使用过多,其中仅生物特性就包括药物的靶标蛋白,转运蛋白和酶等,可能造成信息冗余,产生过学习现象。综上所述,采取机器学习的方法来预测药物副作用是目前研究的一个方向。但算法的改进以及特征的选择一直是药物副作用预测研究的难点和关键问题。技术实现要素:本发明的目的是针对现有技术存在的问题而提供的一种基于多特征融合的药物副作用预测方法,该方法不仅预测精度高,鲁棒性好,而且可以成功预测出一些药物在上市以后才表现出的副作用。该方法可用于药物副作用安全性评估及临床患者用药参考等。实现本发明目的的具体技术方案是:一种基于多特征融合的药物副作用预测方法,特点是该预测方法包括以下具体步骤:步骤1:构建药物特征矩阵a1:计算药物的化学特性相似性所述化学特性相似性为基于药物化学子结构的相似性,化学子结构从pubchem数据库中获得,然后使用式(1)来计算任意两个药物之间的化学特性相似性,得到化学特性相似性矩阵sc;式(1)为药物di和dj之间的化学特性相似性的计算方法;药物的化学子结构对应着881位指纹,structure(d)代表药物化学子结构指纹谱的有效位;a2:计算生物特性相似性生物特性相似性为基于药物靶标蛋白信息的相似性,靶标蛋白从drugbank数据库中获得,然后使用式(2)的计算公式计算任意两个药物之间的生物特性相似性,得到生物特性相似性矩阵sb;式(2)为药物di和dj之间的生物特性相似性的计算方法;t(d)表示药物d的靶标蛋白集合,而|t(d)|表示集合的大小;两个蛋白质之间的序列相似性g(tx,ty)通过smith-waterman序列相似性算法来计算;a3:计算表型特性相似性表型特性相似性为基于药物适应症信息的相似性,药物适应症从sider数据库中获得,两个药物之间的表型特性相似性是通过计算两个适应症在统一医学语言系统中的相似性来获得的,利用bridget等人开发的一个开源软件即可计算两个生物概念条目的路径和语义相似性,得到表型特性相似性矩阵sp;a4:计算药理学特性相似性药理学特性相似性为基于药物atc编码的相似性,atc编码从kegg数据库中获得,部分无法直接获得的,通过在线工具space获取这部分药物的候选atc编码,然后使用式(3)来计算任意两个药物之间的药理学特性相似性,得到药理学特性相似性矩阵st;st(ti,tj)=ω(ti)ω(tj)exp(-γd(ti,tj))(3)式(3)为两个atc编码ti和tj基于一个概率模型来计算药物药理学特性相似性的计算方法;d(ti,tj)表示两个atc编码在atc分类系统的五层层级系统中的最短距离;ω(t)表示相应atc的权重,是atc编码在数据集中出现频率的倒数;γ是预定义参数,设置为0.25;对于有多个atc编码的药物,针对每一个atc编码都计算一个药理学相似性,然后取平均值;步骤2:构建药物副作用的预测模型b1:建模并预测利用k近邻方法分别对化学特性相似性矩阵sc、生物特性相似性矩阵sb和表型特性相似性矩阵sp构建独立的模型,并分别利用这三个独立的模型进行药物副作用预测,得到这三个模型对应的auc分值,aupr分值和运行时间;b2:集成并预测利用集成算法将b1步得到的三个独立的模型进行集成,并利用集成后的模型进行药物副作用预测,得到此模型对应的auc分值,aupr分值和运行时间;b3:建模后集成并预测利用k近邻方法对药理学特性相似性矩阵st建立独立模型,并将b1步得到的三个独立模型与此独立模型集成,利用集成后的模型进行药物副作用预测,得到此模型对应的auc分值,aupr分值和运行时间,以及最终预测结果—药物副作用相互作用关系矩阵。所述集成采用式(4):式中,m表示的是特征的种类;n表示第n个副作用;对于给定新药xi,i表示第i种药物特征数据(i=1,2,…,m);ωi表示第i个特征数据集的相应权重;为一个统计向量,表示的是在药物xi的k个近邻中,存在第n个副作用的药物数目。本发明具有以下优点:1)多样化的特征可能会带来更全面的信息,同时也带来了噪音,问题的关键是如何对特征进行选择。本发明融合了药物的化学特性特征、生物特性特征、表型特性特征和药理学特性特征。特征融合的预测模型可以取得比较好的性能和预测结果,因此,本发明方法对应的auc,aupr值较对比方法更高,运行时间更短。2)新加入的atc编码作为药物药理学特征,使模型比以往的特征融合方法的性能和预测结果均较好。附图说明图1为本发明流程图;图2为本发明与其他三种方法相比较的roc曲线图;图3为本发明与其他三种方法相比较的aupr曲线图;图4为本发明的预测结果统计图。具体实施方式下面结合附图和实施例对本发明进行详细描述。本发明包括:(1)构建药物特征矩阵sider数据库在2015年10月21日发布了最新版本sider4.0,由原来的包含996个药物,4192个副作用以及99423个药物-副作用关系,更新到包含1430个药物和5880个副作用信息以及139756个药物-副作用关系。删除只与一个药物有联系的副作用,经过筛选后,数据集内共有1164个药物对应3795个副作用,以及121348个药物-副作用相互关系。本发明整合了多元异构数据,包含了药物的四个特性,分别是化学特性、生物特性、表型特性和药理学特性。通过计算四个特性特征的相似性,能够进一步为药物副作用预测挖掘特征之间的潜在信息。化学特性相似性使用tanimoto计分法基于药物的化学子结构来计算。药物的化学子结构对应着881位指纹,可以从pubchem数据库中获得。其中在构建的数据集中只有649位是有效位,则两个药物di和dj之间的化学相似性的计算方法如公式(1),其中structure(d)代表药物化学子结构指纹谱的有效位。首先把药物从sider数据库链接到drugbank数据库和stitch数据库,然后收集与药物有联系的靶标,平均每个药物与4个靶标蛋白有联系。我们使用靶标蛋白作为药物的生物特性特征,每个药物因此使用1736维的向量作为特征谱。两个药物di和dj之间的生物特性相似性sb的计算方式如公式(2):其中t(d)表示药物d的靶标蛋白集合,而|t(d)|表示集合的大小。使用smith-waterman序列相似性算法来计算两个蛋白质之间的序列相似性g(tx,ty)。从sider数据库中获取药物的适应症信息作为药物的表型特征,因此每个药物使用2628维的特征向量。两个药物之间的表型相似性sp通过计算两个适应症在统一医学语言系统(unifiedmedicallanguagesystem,umls)中的相似性计算方法来计算。bridget等人开发了一个开源软件umls-similarity,来计算两个生物概念条目的路径和语义相似性。使用药物的atc编码作为药物的药理学特性特征。从sider数据库和kegg数据库来获得药物的atc编码。但仍然有111个药物无法获取atc编码。使用在线工具space(similarity-basedpredictorofatccode)来获取这111个药物的候选atc编码。药物的药理学相似性st通过两个atc编码ti和tj基于一个概率模型来计算,如式(3):st(ti,tj)=ω(ti)ω(tj)exp(-γd(ti,tj))(3)其中d(ti,tj)是两个atc编码在atc分类系统的五层层级系统中的最短距离。ω(t)代表相应atc的权重,是atc编码在数据集中出现频率的倒数。γ是预定义参数,设置为0.25。对于有多个atc编码的药物,针对每一个atc编码都计算一个药理学相似性,然后取平均值。(2)药物副作用预测模型众所周知,药物的特征越相似,药物越有可能有着共同的作用机制,进而有着相同的副作用。因为一个药物经常会与不止一个副作用有关系,因此副作用预测模型更应该是多标签分类问题,而不是传统的二分类{0,1}标量输出问题。本发明方法定义为“多标签集成k近邻方法”(multi-labelensemblek-nearestneighbor,mleknn)。对于一个给定的观测新药,通过观测药物和数据集中的药物之间的相似性来决定距离的远近。整合了多个特征来计算药物之间的相似性,然后使用一个集成优化算法将其结合起来。对于一个给定的新药xi,其中i代表第i种药物特征数据(i=1,2,…,m),则一个统计向量可以被定义为:其中nk代表药物xi在第i种特征相似性上的k个近邻,n代表第n个副作用。模型的不同近邻数的选择对结果会有很大的影响,使用交叉验证来确定最佳近邻数,最优解是k=18。yd(n)=1表示近邻药物d有第n个副作用,相反yd(n)=0表示近邻药物d没有第n个副作用。对于药物的四种特性特征,通过使用平均权重来集成结合四个基于特征的独立模型:其中ωi代表药物第i个特征数据集的相应权重。本发明中,如果整合了c个特征数据集,那么化学子结构,靶标蛋白和适应症都已被证明是有效的预测药物副作用的药物特征。人们提出了几个有效的方法来预测药物副作用,如scca、kr方法及nn方法。使用auc分值,aupr分值和运行时间来评估本发明方法的性能,并且使用其它三种方法作为比较。在统一数据集应用5重交叉验证计算不同特征下本发明方法的性能,不同的特征分别是:化学子结构、靶标蛋白、适应症和三种特征的结合,以及三种特征与atc编码特征的结合。以下通过与scca,kr,nn三种经典方法的比较实验,检测本发明方法预测药物副作用的有效性。比较例1与scca,kr,nn预测的auc分值比较如表i所示,mleknn方法整合了四种特性的异构特征数据时的auc值较其他方法使用其他单一或者三种特征相结合的auc值要略高。每个算法使用特征结合的roc曲线如图2所示,实线代表mleknn方法。表i四种方法的auc分值方法子结构s靶标t适应症i结合(s,t,i)结合(s,t,i,atc)mleknn0.88270.87920.85290.89810.9077scca0.83200.89050.89460.86830.8786kr0.88060.85030.88650.88260.8901nn0.88440.88670.88940.88450.8889比较例2与scca,kr,nn预测的aupr分值比较副作用预测使用的数据集中,正样本的数目要远远小于负样本的数目,因此其aupr分值较其他模型应用略低。如表ii所示,使用mleknn方法结合四种异构特征的aupr值较其他方法高,这说明本发明更稳定并且更具有鲁棒性。每个算法使用特征结合的pr曲线如图3所示,实线代表mleknn方法。表ii四种方法的aupr分值方法子结构s靶标t适应症i结合(s,t,i)结合(s,t,i,atc)mleknn0.33440.30600.30840.36310.4011scca0.29440.30290.32900.32310.3305kr0.30800.30080.32900.32310.3294nn0.32460.32430.33200.32980.3423比较例3与scca,kr,nn预测的运行时间比较与其他方法相比mleknn方法在运行时间上也有大幅度的缩减,如表iii所示,这也是mleknn方法的一个优点:不需要对模型进行学习,因此即使数据量大,但是计算速度快,且可以随时在网络中加入新的药物样本和药物特征。表iii四种方法的运行时间方法子结构s靶标t适应症i结合(s,t,i)结合(s,t,i,atc)mleknn3.073.063.995.787.052scca359.43421.70499.85651.11748.23kr120.86234.95318.65665.41767.65nn266.02279.08353.81349.95356.93(3)预测结果的验证为了对副作用进行一次较综合的预测,通过验证本发明方法的预测结果与药物已知的副作用关系之间的重叠来验证方法的有效性。关注164个药物,其中atc编码分组的比例与我们数据集中的比例相同。检查这164个药物使用本发明方法的预测结果,其统计结果如图4所示。从图4中可以看出,有163个药物的排名前50的预测副作用结果可以被确认。仅有一个药物的副作用结果不能被确认,即磺胺(药物在pubchem数据库中的id是5333)。它仅有两个已知的副作用,并且发生概率均较低(均小于0.01)。因此由于基数小,属于特殊个例,可以近似认为不影响算法模型的预测精度。有40个药物其排名前50个预测结果中的副作用可以被确认超过40个;有86个药物其排名前50的预测结果中的副作用可以被确认超过25个;剩下的药物由于本身表现出的已知的副作用个数较少,不能被确认超过25个,但是仍然有15个药物可以被确认预测结果个数超过已知结果个数的一半。有9个药物其预测结果排名前50的副作用中有49个可以得到确认,这9个药物对应在pubchem中的id,药物名字,药物的适应症以及药物已经表现出的副作用的详细信息如表iv所示。表iv预测副作用确认结果超过49个的药物的详细信息表v药物及其副作用预测结果取药物孕酮(progesterone)为例,其在pubchem数据库中的id是5994,主要作用于子宫,乳腺和大脑,并且作用为中间性腺类固醇激素和肾上腺皮质激素的生物合成。排名前50的预测结果中49个副作用可以得到确认,其中有13个是高表达概率的副作用,如表v所示。其中标“*”的副作用表示这些副作用在药物上市以前都没有表现出来,分别是关节痛(arthralgia)、头晕(dizziness)、头痛(headache)和恶心(nausea)。这里本发明方法都成功的预测出来。吗啡(morphine)在pubchem数据库中的id是4253,是一种镇痛药,其在中枢神经系统和平滑肌中都有广泛的应用。在其预测结果中得到确认的49个副作用中,11个副作用是高表达概率的副作用,如表v所示。其中六个标“*”号的副作用是药物上市以后才表现出的副作用。分别是头晕(dizziness)、颤抖(tremor)、恶心(nausea)、嗜睡(somnolence)、便秘(constipation)和呕吐(vomiting)。沙利度胺(thalidomide)在pubchem数据库中的id是5426,作为一种非巴比妥类催眠药,但由于存在大量的严重的副作用,已从市场撤回。它被报道有342个副作用,其中84个是高表达概率的副作用。在本发明方法的预测的结果中,排名前五十的副作用有49个可以得到确认,其中有28个副作用是高表达概率的副作用(如表v所示)由于大量严重的副作用被成功的预测出来,这进一步证明了mleknn结合多种特征预测药物副作用的有效性和准确性。普罗帕酮(propafenone)在pubchem数据库中的id是4392,对室性心律失常特别有效,其预测结果排名前50的副作用中,得到确认的有47个。其中有22个是高表达概率的副作用,这22个高表达概率的副作用中,标“*”号的17个副作用是在药物上市以后才表现出来,这里本发明方法成功的预测出来了。还有许多药物使用mleknn方法可以得到有意义的预测结果;这些结果暗示着无论是全局准确性还是局部准确性本发明方法都是一个预测药物副作用的有效方法。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1