针对不平衡样本上基因关联分析的误差提升方法与流程
本发明涉及计算生物学技术领域,特别涉及一种针对不平衡样本上基因关联分析的误差提升方法。
背景技术:
目前,基因关联分析(genome-wideassociationstudy,gwas)中的存在一个重要的问题:在分析复杂遗传疾病的时候现有方法缺少足够统计能力来找出所有的致病因素,造成许多潜在的因素没法发现,影响对疾病成因的分析以及诊断。造成这种问题的原因是多样的,而其中一个很容易被忽略的因素为:分析数据中健康样本与患病样本的数目差异。具体来说,在gwas分析中,患病个体的样本是更难采集获得的,因为需要针对患有制定疾病的个体进行采集,例如患胃癌的人群,而与之相对的健康样本可以对健康人群进行随意采集。特别当在稀有疾病上进行gwas分析中,采集患病样本尤其困难。这就造成了分析数据中,患病个体的数据仅仅占了很小的一部分。
在传统的gwas分析过程中,逻辑斯特回归(logisticregression,lr)与χ2统计检验通常用来分析每个基因位点与表现型之间个关系。因为lr可以将其他协变量的因素考虑在内,比如年龄、性别、是否吸烟等,从而被广泛使用。但是,当用lr处理不平衡数据的时候,因为数据健康与患病样本数目这种不平衡关系,导致回归的结果更偏向数目更多的类别,从而在基因序列中隐藏的关联信息强度会被低估,从而减少被发现的能力。
技术实现要素:
本发明旨在至少解决上述技术问题之一。
为此,本发明的目的在于提出一种针对不平衡样本上基因关联分析的误差提升方法,该方法能够显著提高在不平衡数据下的分析能力。
为了实现上述目的,本发明的实施例提出了一种针对不平衡样本上基因关联分析的误差提升方法,包括以下步骤:s1:获取健康样本,并将健康样本随机划分为l个子集,其中,每个子集的样本数目与患病样本的数目相同;s2:将每个健康样本子集与患病样本进行结对,得到l个样本组合,并在每一个样本组合中,挑选出该子集所对应的关键基因位点;s3:根据各个关键基因位点在不同样本组合下发现的次数,对每一个样本组合的重要程度进行得分评估;s4:计算每个健康个体被分配到相应样本组合的得分的均值,并将所述均值作为个体级别的置信分数;s5:对所述每个健康个体的置信分数进行归一化,并将归一化之后得到的分数通过加权逻辑斯特回归进行分析,以检验每一个关键基因位点的统计显著性。
根据本发明实施例的针对不平衡样本上基因关联分析的误差提升方法,通过划分多个平衡样本子集,找出关键基因位点,利用关键基因位点,计算每一个健康个体的重要性权重,最后结合加权逻辑斯特回归,对基因位点对疾病的影响程度进行统计下评定,从而显著提高了在不平衡数据下的分析能力。
另外,根据本发明上述实施例的针对不平衡样本上基因关联分析的误差提升方法还可以具有如下附加的技术特征:
在一些示例中,在所述s2中,挑选关键基因位点的过程包括:从健康样本中随机选择与患病样本数目相同的个体,组建健康样本子集,健康样本子集与患病样本共同组成样本子集对;在样本子集对上,建立基因位点与表现型之间线性回归模型:
其中,ci表示个体第i个基因位点的基因型,y表示个体的表现型;采用lasso进行求稀疏解,得到每个位点的权重wi之后,挑选出前t个最大权重所对应的位点,作为此样本对的关键基因位点。
在一些示例中,所述s4进一步包括:
s41:随机选取不同的健康样本子集,在对应的样本子集对上,建立同样的线性模型,选取对应的t个关键基因位点;
s42:从l个样本子集对上分别挑选出关键基因位点之后,计算出每个基因位点出现在关键基因位点中的频率:
其中,
s43:根据样本子集pl所发现的关键基因位点的得分确定样本子集pl的得分,具体为:
s44:得到每一个样本子集对的得分之后,将每个健康个体的得分作为所对应的样本子集对得分的均值。
在一些示例中,所述s5进一步包括:假设分析样本中有k个健康个体与k个患病样本,每一个健康个体计算得出的样本得分为si(i=1,2,…,k),健康样本归一化得分为:
对于患病个体,其权重定义为:
wi=1/k(i=1,2,…,k),
则加权逻辑斯特回归定义为:
其中,yi表示第i个样本的患病状态,wi为位点的权重,θ为需要估计的权重,xi是回归方程中的协变量。
在一些示例中,所述s5进一步还包括:在估计得到参数之后,显著性检验的统计量定义为:
lr=loglw(θ)-loglw(θ′|null),
其中,loglw(θ′|null)表示不考虑基因位点,仅仅分析协变量的回归结果,2lr服从χ2分布,可以通过对比此分布得到p值。
在一些示例中,在所述s1中,每个子集之间可以存在互相重叠的样本。
本发明的附加方面和优点将在下面的描述中部分给出,部分将从下面的描述中变得明显,或通过本发明的实践了解到。
附图说明
本发明的上述和/或附加的方面和优点从结合下面附图对实施例的描述中将变得明显和容易理解,其中:
图1是根据本发明一个实施例的针对不平衡样本上基因关联分析的误差提升方法的流程图。
图2是根据本发明另一个实施例的针对不平衡样本上基因关联分析的误差提升方法的详细流程示意图。
具体实施方式
下面详细描述本发明的实施例,所述实施例的示例在附图中示出,其中自始至终相同或类似的标号表示相同或类似的元件或具有相同或类似功能的元件。下面通过参考附图描述的实施例是示例性的,仅用于解释本发明,而不能理解为对本发明的限制。
在本发明的描述中,需要理解的是,术语“中心”、“纵向”、“横向”、“上”、“下”、“前”、“后”、“左”、“右”、“竖直”、“水平”、“顶”、“底”、“内”、“外”等指示的方位或位置关系为基于附图所示的方位或位置关系,仅是为了便于描述本发明和简化描述,而不是指示或暗示所指的装置或元件必须具有特定的方位、以特定的方位构造和操作,因此不能理解为对本发明的限制。此外,术语“第一”、“第二”仅用于描述目的,而不能理解为指示或暗示相对重要性。
在本发明的描述中,需要说明的是,除非另有明确的规定和限定,术语“安装”、“相连”、“连接”应做广义理解,例如,可以是固定连接,也可以是可拆卸连接,或一体地连接;可以是机械连接,也可以是电连接;可以是直接相连,也可以通过中间媒介间接相连,可以是两个元件内部的连通。对于本领域的普通技术人员而言,可以具体情况理解上述术语在本发明中的具体含义。
以下结合附图描述根据本发明实施例的针对不平衡样本上基因关联分析的误差提升方法。
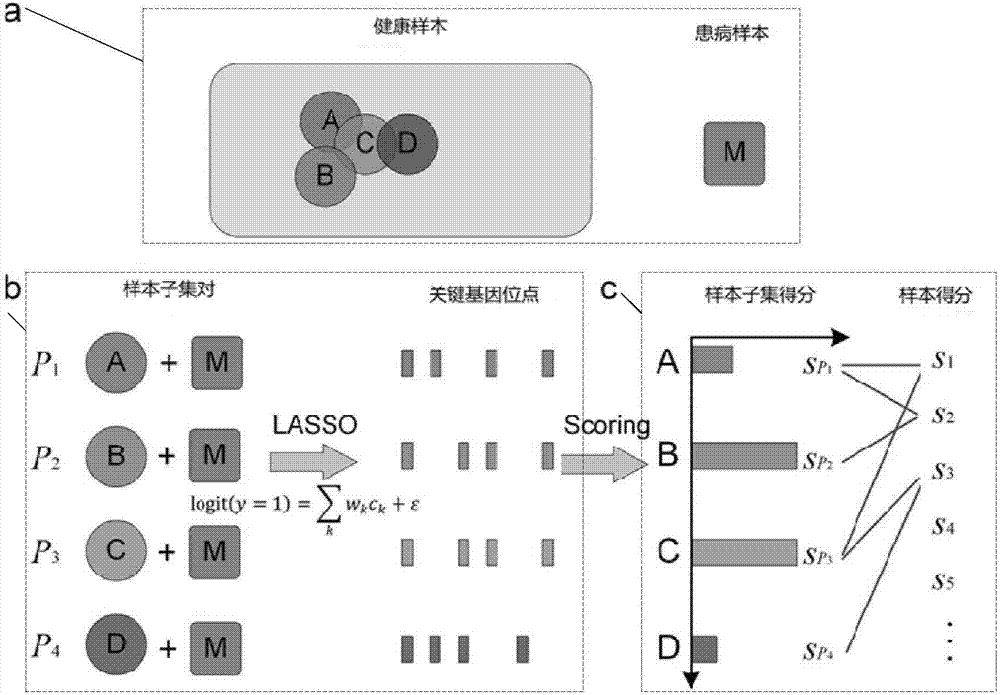
图1是根据本发明一个实施例的针对不平衡样本上基因关联分析的误差提升方法的流程图。图2是根据本发明另一个实施例的针对不平衡样本上基因关联分析的误差提升方法的详细流程示意图。其中,在图2中,a部分展示了所针对的不平衡数据样本示例,以及对健康样本的子集划分方式;b部分展示了样本子集对的组成,通过对样本子集对分析,以及用lasso计算关键基因位点的过程;c部分展示了由关键基因位点计算每个样本子集的得分,以及由样本子集计算样本子集包含的每个样本的得分的过程。
具体地,如图1所示,并结合图2,该方法包括以下步骤:
步骤s1:获取健康样本,并将健康样本随机划分为l个子集,其中,每个子集的样本数目与患病样本的数目完全相同。更为具体地,每个子集之间可以存在互相重叠的样本。
步骤s2:将每个健康样本子集与患病样本进行结对,得到l个样本组合,并在每一个样本组合中,挑选出该子集所对应的关键基因位点。即每个健康样本子集与患病样本进行结对,组成新的分析样本组合。作为具体的示例,例如图2所示,步骤s1中划分的l个子健康样本子集合组成了l个样本组合(p1,...,p4)。进一步地,在每一个组合上面,用稀疏优化方法lasso挑选出子集所对应的关键基因位点(即重要的基因位点)。
在本发明的一个实施例中,挑选关键基因位点的过程包括:首先,从健康样本中随机选择与患病样本数目相同的个体,组建健康样本子集,健康样本子集与患病样本共同组成样本子集对;在样本子集对上,建立基因位点与表现型之间线性回归模型:
其中,ci表示个体第i个基因位点的基因型,y表示个体的表现型;模型假定每个基因位点的对于表现型的作用为线性的,因为每个个体的基因位点特别多,造成方程的欠定。因此,采用lasso进行求稀疏解,得到每个位点的权重wi之后,挑选出前t个最大权重所对应的位点,作为此样本对的关键基因位点。
步骤s3:根据各个关键基因位点在不同样本组合下发现的次数,对每一个样本组合的重要程度进行得分评估。
步骤s4:计算每个健康个体被分配到相应样本组合的得分的均值,并将均值作为个体级别的置信分数。具体地说,每一个健康个体可能被分配到多个样本组合之中,因此,用相应分配所在的样本组合得分的均值作为个体级别的置信分数。
在本发明的一个实施例中,步骤s4进一步包括:
s41:重复上述选择关键基因点的步骤l次,随机选取不同的健康样本子集,在对应的样本子集对上,建立同样的线性模型,选取对应的t个关键基因位点。
s42:从l个样本子集对上分别挑选出关键基因位点之后,计算出每个基因位点出现在关键基因位点中的频率:
其中,
s43:根据样本子集pl所发现的关键基因位点的得分确定样本子集pl的得分,具体为:
s44:得到每一个样本子集对的得分之后,由于每一个健康个体可能会被分配到多个样本子集对之中,因此,将每个健康个体的得分作为所对应的样本子集对得分的均值。
步骤s5:对每个属于健康类别的健康个体的置信分数进行归一化,并将归一化之后得到的分数通过加权逻辑斯特回归进行分析,以检验每一个关键基因位点的统计显著性。
在本发明的一个实施例中,步骤s5进一步包括:假设分析样本中有k个健康个体与k个患病样本,每一个健康个体计算得出的样本得分为si(i=1,2,…,k),健康样本归一化得分为:
对于患病个体,其权重定义为:
wi=1/k(i=1,2,…,k),
则加权逻辑斯特回归定义为:
其中,yi表示第i个样本的患病状态(yi=1为生病,yi=0为健康),wi为位点的权重,θ为需要估计的权重。通过最大似然估计,就可以得到参数的估计结果。xi是回归方程中的协变量,比如性别、年龄等。
进一步地,步骤s5进一步还包括:在估计得到参数之后,进行显著性检验。显著性检验的统计量定义为:
lr=loglw(θ)-loglw(θ′|null),
其中,loglw(θ′|null)表示不考虑基因位点,仅仅分析协变量的回归结果。由于2lr服从χ2分布,因此可以通过对比此分布得到p值。
综上,本发明上述实施例的方法是建立在现有的提升学习理论的基础上,对类别较多的样本加入不同的重要性权重。同时,该方法针对基因数据的特殊结构、进行基因关联分析问题的特殊性进行了专门的设计。整个方法建立在一个“筛选初步-综合分析”的两步学习框架之中。在初步筛选部分,该方法在不同的子数据集下面用一范数约束下的优化lasso选取关键的基因特征。根据在不同子数据集下面提前基因特征的一致性信息,对每一个子数据集进质量评价。在综合分析部分,每个样本的质量评价结果被整合到加权逻辑斯特回归之中,计算每个基因位点级别的统计检验值。
根据本发明实施例的针对不平衡样本上基因关联分析的误差提升方法,通过划分多个平衡样本子集,找出关键基因位点,利用关键基因位点,计算每一个健康个体的重要性权重,最后结合加权逻辑斯特回归,对基因位点对疾病的影响程度进行统计下评定,从而显著提高了在不平衡数据下的分析能力。
在本说明书的描述中,参考术语“一个实施例”、“一些实施例”、“示例”、“具体示例”、或“一些示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不一定指的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任何的一个或多个实施例或示例中以合适的方式结合。
尽管已经示出和描述了本发明的实施例,本领域的普通技术人员可以理解:在不脱离本发明的原理和宗旨的情况下可以对这些实施例进行多种变化、修改、替换和变型,本发明的范围由权利要求及其等同限定。
- 还没有人留言评论。精彩留言会获得点赞!