一种基于动态卷积网络的微生物基因序列分类模型的方法与流程
本发明涉及一种基于动态卷积网络的微生物基因序列分类模型的方法,它应用于微生物基因序列分类识别,属于数据挖掘与生物信息技术领域。
背景技术:
dna序列数据是生物信息学的主要研究对象之一。通过分析dna序列,可以了解序列之间的潜在结构和功能关系。dna序列的数据量呈指数增长,如果可以用现代计算机分析这些巨大的数据来帮助我们了解dna,这是非常好的。dna序列分类遵循具有相似结构的序列也具有相似功能的原理。传统上通过使用序列比对方法(如blast和fasta)建立序列相似性。这个选择是有两个主要假设:(1)功能要素共享共同序列特征,(2)功能元素的相对顺序在不同的间接条件之间是保守的。虽然这些假设在广泛的情况下是有效的,但它们并不普遍。无论如何,尽管最近的这些问题,严重限制对齐方法应用的关键问题仍然是他们计算时间的复杂性。因此,最近开发的无对齐方法已经成为研究基因组分析的有效方法。在无对齐方法中把序列考虑成k-mer集合,然后通过分析每个序列中k-mer分布特性,寻找到有效特征然后用传统的分类方法将序列分类。而基因序列分类中,特征选取和分析常常是费时又费力,而且效果还不确定。
目前,基于深度学习的模型算法在图像识别和自然语言处理等领域中取得了很好的效果,而且越来越受到重视。本方法主要基于深度学习中动态卷积网络实现基于序列的分类。由于深度学习本身会提取高水平抽象特征,从而省去了传统机器学习算法中的特征工程过程,从而极大的提升了问题解决的效率,而且准确度已经达到非常高的水平。
技术实现要素:
1、目的:
本发明目的是提供一种基于动态卷积网络的微生物基因序列分类模型的方法,可以有效的分类微生物基因序列,从而提高微生物分析效率和水平。
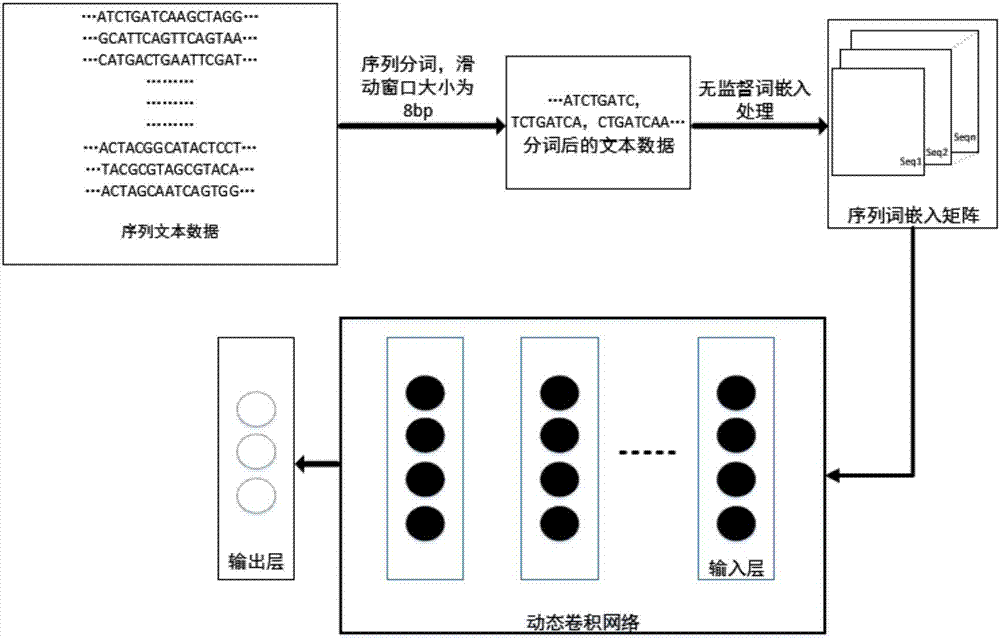
本发明的原理是:首先进行基因序列处理,对某个微生物的基因序列文本进行分词,获取分词结果并作为算法模型输入,算法模型会根据分词结果,首先将序列分词通过词嵌入技术将基因序列转化为向量矩阵,在模型的卷积层,通过一维卷积核对词嵌入矩阵进行卷积,第一层卷积设为12个通道,卷积层后数据进入动态池化层,动态池化层会根据输入序列的长度和当前的卷积层数确定池化域的大小,以最大化保留序列的有效信息。最后在折叠层,把矩阵降维,在全连接层把动态卷积网络提取序列抽象特征进行分类。
2、技术方案:本发明提供的技术方案如下:
本发明是一种基于动态卷积网络的微生物基因序列分类模型的方法,如图2所示,该方法具体步骤如下:
步骤一:获取已有分类结果的微生物基因序列数据。
步骤二:数据预处理:
1)删除基因序列中非法字符;
2)将不同的分类类别属性进行one-hot编码;
3)把基因序列按8个字符切分成词序列;
4)把整理好的序列文本集按对应的分类标签分成门(phylum),纲(class),目(order)科(family),分成四个分类级别数据
步骤三:构建动态卷积网络结构框架,如附图(1);
1)词嵌入层,词嵌入层输入的序列分词向量化。不同的序列词语映射出不同的向量,相近的序列词在映射空间上会更加相近。在此选词向量长度为48;
2)输入层,输入层接收词嵌入层输出的序列向量,然后将数据矩阵传给下一层;
3)卷积层,主要是一维卷积,卷积核扫描输入序列数据矩阵进行卷积运算产生输出结果;所述的卷积层有两个,命名为第一卷积层和第二卷积层
4)动态池化层,池化层根据卷积层数,和输入序列长发动态的选择池化参数,以保留最多有效信息。动态池化层参数根据公式(1)选择:
kl是第l层池化层参数k的选择值,ktop,是最顶层池化层参数,l表示网络中卷积层总层数,l代表是当前的层数,s代表序列的长度选择bp为单位;
5)折叠层,折叠层将所述的第一卷积层和第二卷积层输出矩阵中每上下两层合并,以数值相加等形式进行合并;
6)全连接层,全连接有1024个神经元,可以抽取出神经网络学到的深层次抽象特征;
7)输出层,输出层根据具体分类类别数设置神经元个数,输出神经网络学习的分类结果;
步骤四:把准备好的数据输入步骤三建立的动态卷积网络,用反向传播,随机梯度下降法迭代500次,训练动态卷积网络。以多分类交叉熵为代价函数。最终得到分类算法模型。
步骤五:将序列分词序列输入步骤四已训练好的动态卷积网络模型,输出分类结果。
3、优点及功效:本发明所提供的一种基于动态卷积网络的微生物基因序列分类模型的方法,分类过程不用人工处理数据和抽取特征,模型自动抽取抽象特征完成分类任务,算法效率和准确度都相对较高,可以有效应用于生物信息分析与处理。
附图说明:
图1所示为本发明步骤三建立的动态卷积网络结构图。图中:v1,v2,v3,v4,v5,v6,v7分别代表7个词嵌入向量,输入词向量构成训练数据矩阵,通过卷积层,池化层,折叠层,全连接层抽取出序列抽象特征用以精确分类。
图2所示为本发明方法的流程图。
具体实施方式:
下面结合附图和实施例,对本发明的技术方案做进一步的说明。
本发明一种基于动态卷积网络的微生物基因序列分类模型的方法,具体包括如下步骤:
步骤一:获取微生物基因序列数据(以rdprrna基因数据为例下载地址为:http://rdp.cme.msu.edu/http://rdp.cme.msu.edu/.此数据有已标记3,356,809条微生物16srrnas数据),从中随机采样30万条序列数据,用以之后的模型训练。
步骤二:数据预处理:
1)删除基因序列中非法字符;
2)将不同的分类类别属性one-hot编码;
3)把基因序列按8个字符切分成词序列;
例如:gtttataagggcttgccctt端序列:可以将其分词为:gtttataa,tttataag,ttataaggtataaggg,ataagggc,taagggct,aagggctt,每8个bp看做一个词语。
如以下序列所示:
gtttataagggcttgcccttatagatagtggcgaacgggtgcgtaacacgtgagcaacctgccccaaagtttggaataacaccgggaaaccgatgctaataccaaatatgctcacactatcacaagatagagtgaggaaagtttttcgctttgggaggggctcgcggcctatcagcttgttggtgaggtaacggctcaccaaggcatcgacgggtagctggtctgagaggacgatcagccacactgggactgagacacggcccagactcctacgggaggcagcagtggggaatattgcgcaatgggcgaaagcctgacgcagcaacgccgcgtggaggatgaaggccttagggtcgtaaactcctttcagcaggaacgaaaatgacggtacctgcagaagaagctccggccaactacgtgccagcagccgcggtaatacgtagggagcaagcgttgtccggatttattgggcgtaaagagctcgtaggcggcttggcaagtcggatgtgaaacccccaggcttaacctggggccgccattcgatactgctatggcttgagttcggtaggggattgtggaattcccggtgtagcggtgaaatgcgcagatatcgggaggaac将其按每8个字符拆分,得到以下序列文本集:
4)把整理好的序列文本集按对应的分类标签分成门,纲,目,科,分成四个分类级别数据;
步骤三:构建动态卷积网络结构框架,网络结构如图附图(1);对网络参数设置有ktop=5,词嵌入向量长度为48,第一层卷积设为12个通道,第二层卷积设为8个通道,输出层神经元个数根据不同的分类任务类别数而定,例如本次实验phylum:50,class:114,order:256,family:1386。
步骤四:把准备好的数据输入步骤三建立的动态卷积网络,用反向传播,随机梯度下降法迭代500次,每次选择batchsize为200个训练样本训练动态卷积网络。以多分类交叉熵为代价函数。最终得到分类算法模型。实验表示在模型迭代500次左右基本收敛,分类准确率能达到99.6%左右,较之传统机器学习算法有很大提升。
步骤五:将需要分类的分词序列输入到步骤五已训练好的动态卷积网络模型,到分类如下结果。
phylum:actinobacteria
class:actinobacteria
order:acidimicrobiales
family:acidimicrobiaceae。
- 还没有人留言评论。精彩留言会获得点赞!