一种计算机模拟获取G蛋白偶联受体中间态结构的方法与流程
本发明属于g蛋白偶联受体结构的计算机模拟技术领域,具体涉及一种计算机模拟获取g蛋白偶联受体中间态结构的方法。
背景技术:
g蛋白偶联受体(gpcr)是人体中最大的膜蛋白家族之一,是一类重要的药物靶标,市场上约30%的药物以gpcr为药物靶标,因此对其结构和功能研究已成为国际的研究热点。由于gpcr是一种膜蛋白,实验手段对其结构解析非常困难,目前通过x射线结晶等实验技术获取的受体结构非常有限,且大多数是稳定的非活性的静态结构,仅有少量的活性态结构解析出来,但是依靠这些实验结晶的静态结构不能有效获取其发挥信号功能的动态过程。gpcr在激活过程中伴随一系列大的结构重排,在受体结构重排中会产生重要的中间态,这些中间态在其信号传导中有着重要的功能,但是这些中间态存在的时间非常短,采用实验上的技术方法难以捕获,这些中间态结构的缺失明显限制了gpcr结构和功能的研究,使得目前对gpcr激活机制的阐明以及药物设计存在较大困难。
计算机模拟方法(分子动力学模拟)可以在原子水平上获取极快时间内蛋白功能变化过程中的微观结构,刚好可以弥补这些实验上的不足,但是gpcr激活过程发生在毫秒和秒级的时间范围,采用常规分子动力学模拟(cmd)的方法(流程如附图1所示)在目前的计算机资源上很难达到此时间尺度,难以获得变化过程中的所有结构。
技术实现要素:
有鉴于此,本发明的目的在于提供一种结合靶向分子动力学和常规分子动力学模拟的方法来突破计算模拟的时间限制,更有效地获取gpcr从非活性态到活性态的激活过程中重要的中间态结构的方法。
为了实现上述发明目的,本发明提供以下技术方案:
一种计算机模拟获取g蛋白偶联受体中间态结构的方法,包括以下步骤:1)去掉g蛋白偶联受体非激活和激活态晶体结构的非受体分子,获得无配体状态的受体;2)在charmm-gui或vmd软件中构建模拟体系,所述模拟体系中包括无配体状态的受体、磷脂双分子层、溶剂分子和离子;3)在分子动力学模拟软件中的动力学模拟模块中,采用最陡下降法和共轭梯度法对所述模拟体系进行限制性能量最小化的优化获得优化模拟体系;4)使用berendsen温度耦合法对所述优化模拟体系进行升温,升温过程中对所述优化模拟体系中蛋白质的重原子进行位置限制,获得二次优化模拟体系;5)在分子动力学模拟软件中的动力学模拟模块中,采用npt系综,在所述二次优化模拟体系中进行靶向分子动力学模拟获得tmd轨迹;6)在分子动力学模拟软件中的分析模块中对获得的tmd轨迹进行聚类分析,选取每类的中心作为cmd模拟的初始结构;7)对cmd模拟的若干初始结构进行常规分子动力学模拟,获得cmd轨迹;8)对获得的cmd轨迹进行聚类分析,根据聚类分析的结果选择确定g蛋白偶联受体中间态结构。
优选的,步骤1)中所述g蛋白偶联受体非激活和激活态晶体结构的来源为pdb数据库。
优选的,步骤3)所述的限制性能量最小化优化包括依次进行的溶剂分子的结构和位置优化、溶剂分子和受体蛋白结构的位置优化,受体蛋白侧链分子优化和其余结构优化。
优选的,步骤3)中每阶段优化的步数为2000~20000步,其中先采用最陡下降法去松弛过接近的连接,后采用共轭梯度法。
优选的,步骤4)中所述升温为将优化模拟体系的温度在150~200ps的时间内由0k上升到300~310k。
优选的,步骤4)中所述蛋白质的重原子位置限制权重值为
优选的,步骤5)中所述靶向分子动力学模拟以g蛋白偶联受体非激活和激活态的晶体结构分别作为模拟的起始态和终点态。
优选的,步骤5)中所述npt系综的模拟温度为300~310k,压力为1个大气压的周期边界条件,其中与h相连的键长,限制的阈值为
优选的,步骤5)中所述的tmd模拟步长为1~2fs,每隔0.2~2ps保存轨迹。
优选的,步骤6)和步骤8)中所述聚类分析采用k-means算法基于氨基酸残基的骨架原子进行聚类分析。
本发明的有益效果:本发明所述的计算机模拟获取g蛋白偶联受体中间态结构的方法,利用靶向分子动力学(tmd)模拟来克服常规分子动力学(cmd)模拟时间的局限性,初步获取gpcr从非活性态到活性态的激活过程,并通过聚类分析获取激活过程的中间态cmd初始结构,然后再采用cmd模拟的方法对这些中间态cmd初始结构进行进一步的模拟,汇集中间态的cmd模拟轨迹获取其激活过程的构象变化空间,然后对其轨迹进行聚类分析,通过构象的布局数和结构差异,获取gpcr从非活性态到活性态的激活过程中的中间态结构。本发明所述方法可以解决实验技术对gpcr激活过程中间态结构的解析难题,同时解决常规全原子分子动力学模拟在毫秒以及秒级时间尺度内的模拟时间限制的问题。本发明所述方法适用范围广,应用于原子数目较多的大体系以及原子数目较少的小体系;本发明所述的方法简单易行,在计算机中安装相应的软件,根据操作流程进行模拟即可,不必进行复杂的实验操作;本发明所述的方法节约时间和计算机资源,tmd可以在短时间内模拟出研究体系由起始态到终态的运动轨迹,大大减少了模拟时间也降低了对计算机性能的要求;相对于实验上的宏观观测,本发明便于从原子水平来研究体系的运动变化,并且可以克服由起始态到终态的模拟时间问题,观测到两个态之间的整个变化过程。
附图说明
图1为现有技术中分子动力学模拟的流程示意图;
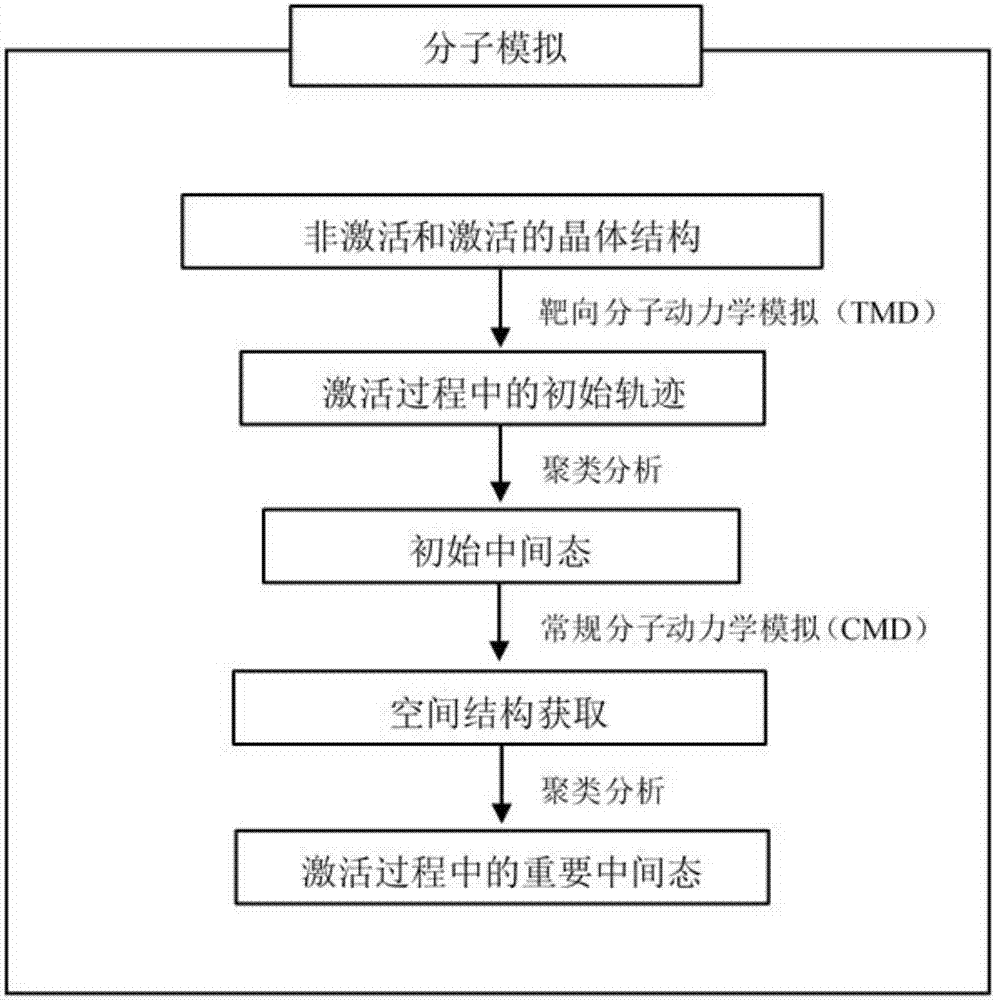
图2为本发明所述方法模拟获得gpcr中间态结构的流程示意图;
图3为实例2获得的3个重要的中间态的结构图。
具体实施方式
本发明提供了计算机模拟获取g蛋白偶联受体中间态结构的方法,包括以下步骤:1)去掉g蛋白偶联受体非激活和激活态晶体结构的非受体分子,获得无配体状态的受体;2)在charmm-gui或vmd软件中构建模拟体系,所述模拟体系中包括无配体状态的受体、磷脂双分子层、水和离子;3)在分子动力学模拟软件中的动力学模拟模块中,采用最陡下降法和共轭梯度法对所述模拟体系进行限制性能量最小化的优化获得优化模拟体系;4)使用berendsen温度耦合法对所述优化模拟体系进行升温,升温过程中对所述优化模拟体系中蛋白质的重原子进行位置限制,获得二次优化模拟体系;5)在分子动力学模拟软件中的动力学模拟模块中,采用npt系综,在所述二次优化模拟体系中进行靶向分子动力学模拟获得tmd轨迹;6)在分子动力学模拟软件中的分析模块中,对获得的tmd轨迹进行聚类分析,选取每类的中心作为cmd模拟的初始结构;7)对若干初始结构进行常规分子动力学模拟,获得cmd轨迹;8)对获得的cmd轨迹进行聚类分析,根据聚类分析的结果选择确定g蛋白偶联受体中间态结构。
本发明所述方法所用工具软件为分子动力学模拟软件,本发明对所述分子动力学模拟软件的种类没有限定,只要能够实现分子动力学模拟即可,在本发明实施过程中优选的采用amber16计算软件;本发明所述方法利用上述软件对g蛋白偶联受体进行一种tmd和cmd组合的分子动力学模拟。
本发明所述的方法流程如附图2所示,首先采用靶向分子动力学方法(tmd)克服激活过程的高能垒,获取g蛋白偶联受体从非活性到活性态激活过程的初始中间态结构,然后基于所述中间态结构进一步进行常规分子动力学模拟,整合获取gpcr激活过程的轨迹,再采用聚类分析获得激活过程中间态的结构,利于对中间态的进行深入研究,进一步阐明gpcr激活机制,也为以gpcr为靶标的药物设计提供新思路。
在本发明中,所述g蛋白偶联受体非激活和激活态晶体结构的数据来源于pdb数据库,从pdb数据库下载需要获得中间态结构的g蛋白偶联受体的非激活和激活态晶体结构。
本发明中,去掉g蛋白偶联受体非激活和激活态晶体结构的非受体分子,获得无配体状态的受体。本发明对所述去掉g蛋白偶联受体非激活和激活态晶体结构的非受体分子的方法无特殊限定,采用本领域的常规方法即可,在本发明具体实施过程中优选的采用pymol软件。本发明中去掉非受体分子的目的是确保非激活和激活态结构初始结构原子数目的类型一致,利于后续模拟。
本发明在charmm-gui(http://www.charmm-gui.org/)或vmd软件中构建模拟体系;所述模拟体系包括无配体状态的受体、磷脂双分子层、水和离子;对于离子的选择,根据不同gpcr受体所带的电荷进行调节,可加入单一的抗衡离子,如na+,k+或cl-中的一种将体系保持为电中性,也可加入不同配比的na+/cl-或k+/cl-提供一定的离子强度,并保持体系呈现电中性。所述磷脂双分子层、水和离子的数量比例为优选的为(10~40):1000:(16~18)。在本发明中所述构建获得的模拟体系存在很多不自然的接触,体系势能高。
本发明为了消除模拟体系中体系势能高、原子位子不合理的情况,在分子动力学模拟软件中的动力学模拟模块中,采用最陡下降法和共轭梯度法对模拟体系进行限制性能量最小化的优化获得优化模拟体系。所述分子动力学模拟软件优选的为amber16;所述动力学模拟模块优选的为sander模块。在本发明中所述限制性能量最小化优化包括依次进行的溶剂分子的结构和位置优化、溶剂分子和受体蛋白结构的位置优化,受体蛋白侧链分子优化和其余结构优化。在本发明中所述限制性能量最小化的优化中受体蛋白是由ff14sb力场描述,磷脂分子由gaff(generalamberforcefiled)力场描述。在本发明中所述溶剂分子优选的为水,所述溶剂分子的结构和位置优化的过程中,采用
本发明在获得优化模拟体系后,使用berendsen温度耦合法对所述优化模拟体系进行升温,升温过程中对蛋白质的重原子进行位置限制,获得二次优化模拟体系。在本发明中,所述升温为将优化模拟体系的温度在150-200ps的时间内温度由0k升温到300-310k,其中所述蛋白质的重原子位置限制权重值优选的为
本发明在获得二次优化模拟体系后,分子动力学模拟软件中的动力学模拟模块中,采用npt系综,在二次优化模拟体系中进行靶向分子动力学模拟,获得tmd轨迹。在本发明中,靶向分子动力学模拟采用的是npt系综,优选的在amber16程序的sander模块中实现。tmd模拟的起始和终点坐标分别是上述所得g蛋白偶联受体非激活和激活态的晶体结构。优选的,npt系综的模拟温度为310k,压力为1个大气压的周期边界条件,温度控制优选的采用了berendsen温度耦合法,压力控制优选的采用基于分子的各向同性校正法。shake算法优选的被用来限制所有与h相连的键长,其中限制的阈值为
在本发明中,为了探究最佳的tmd模拟条件,优选的测试限制力范围为
本发明在获得tmd轨迹后,分子动力学模拟软件中的分析模块中对获得的tmd轨迹进行聚类分析,选取每类的中心作为cmd模拟的初始结构。优选的在amber16的cpptraj模块中。在本发明中,为了获取更为真实的受体构象,优选的采用上述获得的若干条tmd轨迹中最优的一条进行聚类分析,最优的模拟轨迹的起始点和终点分别与采用的起始态和终点态的晶体结构最接近。在本发明中所述聚类分析优选的采用k-means算法基于氨基酸残基的骨架原子进行。根据轨迹中保存的构象数目在2~200类的范围内选取若干组作聚类测试,根据聚类结果中的dbi值(davies-bouldinindex)以及每一类中所有构象的rmsd值(root-mean-squaredeviation,均方根偏差)来确定最终的聚类数目。所述dbi值优选的采用最小的,所述的均方根偏差的差异至少小于
本发明在获得cmd模拟的初始结构后,对若干初始结构进行常规分子动力学模拟,获得cmd轨迹。在本发明中所述常规分子动力学模拟的条件参数与上述tmd模拟的条件参数一致,在此不再赘述。在本发明中,cmd模拟过程中,每个初始结构模拟一段时长,所述一段时长优选的为150~1000ns,更优选的为300~800ns。本发明通过cmd模拟获得若干条cmd模拟轨迹。
本发明在获得cmd模拟轨迹后,对获得的cmd轨迹进行聚类分析,根据聚类分析的结果确定g蛋白偶联受体中间态结构。在本发明中所述聚类分析优选的采用k-means算法基于氨基酸残基的骨架原子进行。本发明根据聚类结果的dbi值(davies-bouldinindex)以及每一类中所有构象的rmsd值来设定最合理的类别数目,所述dbi值优选的采用最小的,所述的均方根偏差的差异至少小于
下面结合实施例对本发明提供的计算机模拟获取g蛋白偶联受体中间态结构的方法进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
实施例1计算机模拟获取肾上腺素受体(β2ar)中间态结构
①从pdb数据库下载肾上腺素受体(β2ar)非激活和激活态的晶体结构,pdbid号分别是2rh1和3sn6。
②为了确保非激活和激活态结构初始结构原子数目的类型一致,在pymol软件中去掉所有的非受体分子,最后得到无配体状态的受体(apo)
③在charmm-gui(http://www.charmm-gui.org/)中加入磷脂双分子层(popc)、水、离子等构建受体需要的模拟环境。
④随机构建的体系可能存在很多不自然的接触,体系势能会很高,所以为了消除模拟体系中原子位子不合理的情况,在amber16的sander模块中,在体系恒温的情况下采用最陡下降法对体系进行4次限制性能量最小化的优化,每次步长均为20000步。其中第一次优化时,采用
⑤完成优化后,继续在amber16的sander模块中使用berendsen温度耦合法,使体系的温度在150ps的时间内以每10ps升温20k的速度由0k上升到300k,分15次完成升温过程。在升温过程中,对蛋白质的重原子进行位置限制,权重值为
⑥加热结束后,进一步对模拟体系进行平衡。tmd模拟采用的是npt系综,仍然在amber16程序的sander模块中实现。tmd模拟的起始和终点坐标分别是上面所得非激活态和激活态的坐标。此外,npt系综的模拟温度为310k,压力为1个大气压的周期边界条件,温度控制采用了berendsen温度耦合法,压力控制采用基于分子的各向同性校正法。shake算法被用来限制所有与h相连的键长,其中限制的阈值为
⑦为了获取更为真实的受体构象,选取第6步中限制力为
⑧对从tmd轨迹上得到的6个起始结构进行cmd模拟,模拟条件见步骤6,每个起始结构模拟150ns;
⑨合并步骤8得到的6轨迹,同样采用k-means算法基于氨基酸残基的骨架原子对总长900ns的cmd轨迹进行聚类分析,根据聚类结果的bdi值为1.4以及每类所有构象的rmsd值差异在0.5左右设定最合理的类别数目为60类,而根据每类的构象数目以及结构特征,最终选取其中最具代表性的10类作为受体激活过程的中间态结构,结果如表1所示,10个重要中间态在配体结合区和g蛋白结合区的各个区域具有不同的结构特征,也具有不同的活性。
表1为得到的10个重要中间态在各区域的不同结构特征,其中,conformationstates即构象名称;rmsd_all代表全原子的rmsd(root-mean-squaredeviation,均方根偏差),其中ina代表以非活性态(inactive)晶体结构为参照,act代表以活性态(active)晶体结构为参照;volume为配体结合口袋体积;d_207-316代表残基207与316的距离(distance);rmsd_ecl2代表ecl2区的rmsd值;d_193-308代表残基193与308的距离(distance);d_tm3-tm6代表螺旋3与螺旋6的距离;d_ionic代表离子锁的距离;rmsd_npxxy代表npxxy区的rmsd值;rmsd_icl2为icl2区域的rmsd值。
表110个重要中间态在各区域的不同结构特征
实施例2计算机模拟获取μ-阿片受体(μ-opioid)中间态结构
①从pdb数据库下载μ-阿片受体(μ-opioid)非激活和激活态的晶体结构,pdbid号分别是4dkl和5c1m。
②为了确保非激活和激活态结构初始结构原子数目的类型一致,在pymol软件中去掉所有的非受体分子,最后得到无配体状态的受体(apo)
③在charmm-gui(http://www.charmm-gui.org/)中加入磷脂双分子层(popc)、水、离子等构建受体需要的模拟环境。
④随机构建的体系可能存在很多不自然的接触,体系势能会很高,所以为了消除模拟体系中原子位子不合理的情况,在amber16的sander模块中,在体系恒温的情况下采用最陡下降法对体系进行4次限制性能量最小化的优化,每次步长均为20000步。其中第一次优化时,采用
⑤完成优化后,继续在amber16的sander模块中使用berendsen温度耦合法,使体系的温度在200ps的时间内以每10ps升温15.5k的速度由0k上升到310k,分20次完成升温过程。在升温过程中,对蛋白质的重原子进行位置限制,权重值为
⑥加热结束后,进一步对模拟体系进行平衡。tmd模拟采用的是npt系综,仍然在amber16程序的sander模块中实现。tmd模拟的起始和终点坐标分别是上面所得非激活态和激活态的坐标。此外,npt系综的模拟温度为310k,压力为1个大气压的周期边界条件,温度控制采用了berendsen温度耦合法,压力控制采用基于分子的各向同性校正法。shake算法被用来限制所有与h相连的键长,其中限制的阈值为
⑦为了获取更为真实的受体构象,选取第6步中限制力为
⑧对从tmd轨迹上得到的2个起始结构进行cmd模拟,模拟条件见步骤6,每个起始结构模拟450ns;
⑨合并得到的几条cmd轨迹,同样采用k-means算法基于氨基酸残基的骨架原子对总长900ns的cmd轨迹进行聚类分析,根据聚类结果的bdi值为1.3以及每类所有构象的rmsd值差异在0.4左右设定最合理的类别数目为10类,而根据每类的构象数目以及结构特征,最终选取其中最具代表性的3类作为受体激活过程的中间态结构,结果如图3所示,其中inactivestate为非活性状态,intermediatestate为中间活性状态,activestate为活性状态;受体的上方为配体结合区域,受体下方为g蛋白结合区域。3个中间态在配体结合区具有不同的口袋形状和大小,而在g蛋白结合区的tm6(跨膜螺旋6)具有不同移动变化,不同的结构特征也就导致中间体具有不同的活性和功能。
由以上实施例可知,本发明所述方法利用amber16计算软件进行一种组合的分子动力学模拟。首先在pdb数据库中下载gpcr非活性和活性态的晶体结构,先采用靶向分子动力学方法突破激活过程的高能垒,获取gpcr从非活性到活性态激活过程的中间态构象种子,然后基于这些初始的中间态种子进一步进行常规分子动力学模拟,整合获取gpcr激活过程的轨迹,并采用聚类分析获得激活过程中间态的结构,方便对中间态的深入研究。该方法可以解决实验技术对gpcr激活过程中间态结构的解析难题,同时解决常规全原子分子动力学模拟在毫秒以及秒级时间尺度内的模拟时间限制。该方法对计算机设备要求低,简便易行,且应用范围广,可以应用在生物、医药、化学等领域从原子水平上研究体系的运动过程以及中间体结构的解析。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!
- 5‑HT2B拮抗剂的制造方法与工艺
- 1‑(噻唑‑2‑基)哌啶‑4‑醇的合成方法与制造工艺
- 一种大麻酚类化合物的合成方法与制造工艺
- 一类对GPR84具有激动作用的化合物及其制备方法和用途与制造工艺
- eGFP标记GPR120阳性细胞的小鼠模型构建方法
- 靶向生长抑素受体的Al<sup>18</sup>F?NOTA?PEG<sub>6</sub>?TATE及其制备方法和应用
- 吡咯烷衍生物的口服制剂的制作方法
- 胰高血糖素样肽-1受体激动剂制备治疗肺动脉高压药物的应用
- 一种armc5基因检测试剂盒及突变位点的制作方法
- 作为mglu5受体的负性别构调节剂的6-氟-2-[4-(吡啶-2-基)丁-3-炔-1-基]咪唑并[1,2a ...的制作方法