一种基于改进L1正则化和聚类的高维数据特征选择方法
本发明涉及机器学习技术领域,尤其涉及一种基于改进l1正则化和聚类的高维数据特征选择方法。
背景技术:
临床上已证实很多疾病同基因之间存在着密切的关系。通常,表达水平与疾病发生高度相关的基因被称为生物标志物,生物标志物的发现对于疾病的早期诊断和预防具有重要意义。为寻找信息最丰富的生物标志物并去除冗余和与目标疾病不相关的生物标志物,微阵列数据分析技术应运而生。
微阵列数据分析技术用于确定生物标志物。众所周知,原始微阵列数据中与疾病相关的特征(基因)的实际数量相对较少,这是由于特征维度高和样本量小所致。这类数据通常包含少量样本和大量与目标疾病无关的特征。另外,微阵列数据具有很高的复杂性,即特征是具有高度冗余性的直接或相互关联的结果,这使得许多应用的机器学习算法显示出低鲁棒性和差的分类精度。因此,在构建模型之前寻找一种合适的方法来减少特征数量,提高模型的分类精度和鲁棒性具有十分重要的意义。
特征选择对于挖掘大规模高维数据集,如微阵列和质谱分析生成的数据集,建立统计模型具有重要意义。在特征选择中,可以识别整个训练数据集中的显着特征。特征选择是在高维,小样本的生物数据中选择生物标记的中的重要一步。常见的特征选择方法可以分为过滤法、包装法和嵌入法,而目前较为先进的特征选择方法是将三种方法进行不同方式的改进和组合而成的混合式特征选择方法。这些方法大多采用叠加两个以上的特征选择方法,用来提高分类准确率。然而在微阵列数据分析中,研究人员往往更加关注特征选择结果的稳定性和特征子集间的非冗余性,即特征子集之间存在的冗余关系较少。
l1正则化是机器学习中的重要手段,通过对代价函数中增加l1范数作为惩罚项来实现稀疏系数矩阵,实现特征选择的目的,而改进的l1正则化方法基于抽样和选择相结合,弱化了特征选择结果对于正则化系数的敏感性,能够显著提升结果稳定性,控制假阳性。聚类是一个将数据集中在某些方面相似的数据成员进行分类组织的过程,k-means聚类算法能够通过基于欧氏距离的计算方法,将样本划分为若干个关联较弱的子集,能够实现特征的聚类和筛选。
技术实现要素:
针对现有技术的不足,本发明提供一种基于改进l1正则化和聚类的高维数据特征选择方法。
本发明的技术方案为,一种基于改进l1正则化和聚类的高维数据特征选择方法,包括以下步骤:
步骤1:根据给定的基因微阵列数据集,利用k-means聚类算法实现基因微阵列数据特征的聚类;
步骤1.1:以基因微阵列数据样本集d={x1,x2,…,xm}为输入,进行k-means聚类算法,其中聚类簇数量k,xj代表样本集中第j个特征,m为样本数目;
步骤1.2:从样本集d中随机选择k个样本作为初始均值向量{μ1,μ2,…,μk},其中μi代表第i个样本对应的均值向量;
步骤1.3:对于样本集d中每一个特征xj,初始化令j=1,执行如下操作:
步骤1.3.1:定义存储样本经过聚类后对应的簇
步骤1.3.2:计算特征xj与每一个均值向量μi的距离,并记为dji,公式如下所示;
dij=||xj-μi||2(1)
步骤1.3.3:计算特征xj的簇标记λj,公式如下所示;
步骤1.3.4:将特征xj放入到对应的簇中,即
步骤1.3.5:令j=j+1,判断j是否大于m,若大于转到步骤1.4,否则转到步骤1.3.2;
步骤1.4:对于每一个均值向量μi,令i=1,执行如下操作:
步骤1.4.1:对μi的值进行更新并记为μ′i,如下式所示;
其中x表示所有数据集合ci的特征;
步骤1.4.2:判断当前μi是否等于μ′i,若是则转到步骤1.4.3,否则保持当前μi不变,转到步骤1.4.4;
步骤1.4.3:将当前均值向量μi的值更新为μ′i;
步骤1.4.4:令i=i+1,并判断i是否大于k,若是则转到步骤1.5,否则转到步骤1.4.1;
步骤1.5:若当前均值向量μi被更新,则转到步骤1.3,否则转到步骤1.6;
步骤1.6:对于得到的所有的ci,其中i=1,2,…,k,令c={c1,c2,…,ck};
步骤1.7:输出划分之后的簇c={c1,c2,…,ck};
步骤2:对于步骤1中产生的每个簇c1-ck,利用皮尔森相关系数迭代删除冗余特征,更新每个簇;
步骤2.1:对于划分之后的簇c={c1,c2,…,ck},令参数q=1,执行以下步骤:
步骤2.1.1:对于cq计算每个特征xi的独立样本t检验统计量p值,公式如下所示;
其中
步骤2.1.2:对所有
步骤2.1.3:计算簇cq中除种子节点xs外所有节点与xs的相关系数
其中e为数学期望;
步骤2.1.4:将相关系数按照从大到小排序,删除每个簇中前15%相关系数对应的节点;
步骤2.1.5:保留余下的节点作为新的簇
步骤2.1.6:令q=q+1,判断q是否大于k,如果是转到步骤2.2,否则转到步骤2.1.1;
步骤2.2:令更新后的簇集合
步骤2.2.1:利用改进l1正则化的特征选择算法,对输入的每个簇
步骤2.2.1.1:输入样本空间
步骤2.2.1.2:在样本空间x中随机采样
步骤2.2.1.3:用lasso回归模型计算损失函数e(x*,y*)+α‖w‖,其中w为惩罚项系数。
步骤2.2.1.4:将回归模型中xi对应的系数记为t,则证明该特征被选择,令其特征权重
步骤2.2.1.5:令i=i+1,判断i是否大于k,若是则转到步骤2.2.1.6,否则转到步骤2.2.1.2.
步骤2.2.1.6:输出所有xi对应的特征权重
步骤2.2.2:令w=w+1,判断w是否大于k,若是则执行步骤2.3,否则执行步骤2.2.1;
步骤2.3:计算每个特征的累加权重
步骤2.4:根据pw的排序结果,输出前l个特征作为最终的特征集合f={f1,f2,…,f},其中f1对应pw中累加权重最大的一项;
步骤3:对于最终得到的特征集合f={f1,f2,…,fl},从原始微阵列数据中找到对应的基因名称,完成对基因的特征分析。
采用上述技术方法所产生的有益效果在于:
本发明提供一种基于改进l1正则化和聚类的高维数据特征选择方法,提出了一种混合特征选择算法用于微阵列数据分析,基于k-means聚类算法和改进l1正则化的思想,其中k-means聚类算法用于数据预处理来删除冗余特征,改进l1正则化方法用于特征选择,提高稳定性和分类准确率。
附图说明
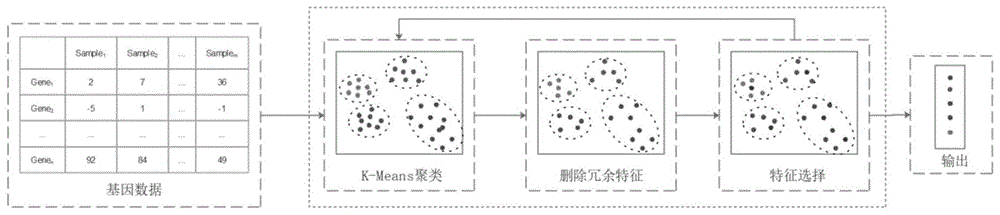
图1为本发明整体流程图。
具体实施方式
下面结合附图和实施例,对本发明的具体实施方式作进一步详细描述。以下实施例用于说明本发明,但不用来限制本发明的范围。
一种基于改进l1正则化和聚类的高维数据特征选择方法,如图1所示,包括以下步骤:
步骤1:根据给定的基因微阵列数据集,利用k-means聚类算法实现基因微阵列数据特征的聚类;
步骤1.1:以基因微阵列数据样本集d={x1,x2,…,xm}为输入,进行k-means聚类算法,其中聚类簇数量k,xj代表样本集中第j个特征,m为样本数目;
步骤1.2:从样本集d中随机选择k个样本作为初始均值向量{μ1,μ2,…,μk},其中μi代表第i个样本对应的均值向量;
步骤1.3:对于样本集d中每一个特征xj,初始化令j=1,执行如下操作:
步骤1.3.1:定义存储样本经过聚类后对应的簇
步骤1.3.2:计算特征xj与每一个均值向量μi的距离,并记为dji,公式如下所示;
dji=||xj-μi||2(1)
步骤1.3.3:计算特征xj的簇标记λj,公式如下所示;
步骤1.3.4:将特征xj放入到对应的簇中,即
步骤1.3.5:令j=j+1,判断j是否大于m,若大于转到步骤1.4,否则转到步骤1.3.2;
步骤1.4:对于每一个均值向量μi,令i=1,执行如下操作:
步骤1.4.1:对μi的值进行更新并记为μ′i,如下式所示;
其中x表示所有数据集合ci的特征;
步骤1.4.2:判断当前μi是否等于μ′i,若是则转到步骤1.4.3,否则保持当前μi不变,转到步骤1.4.4;
步骤1.4.3:将当前均值向量μi的值更新为μ′i;
步骤1.4.4:令i=i+1,并判断i是否大于k,若是则转到步骤1.5,否则转到步骤1.4.1;
步骤1.5:若当前均值向量μi被更新,则转到步骤1.3,否则转到步骤1.6;
步骤1.6:对于得到的所有的ci,其中i=1,2,…,k,令c={c1,c2,…,ck};
步骤1.7:输出划分之后的簇c={c1,c2,…,ck};
步骤2:对于步骤1中产生的每个簇c1-ck,利用皮尔森相关系数迭代删除冗余特征,更新每个簇;
步骤2.1:对于划分之后的簇c={c1,c2,…,ck},令参数q=1,执行以下步骤:
步骤2.1.1:对于cq计算每个特征xi的独立样本t检验统计量p值,公式如下所示;
其中
步骤2.1.2:对所有
步骤2.1.3:计算簇cq中除种子节点xs外所有节点与xs的相关系数
其中e为数学期望;
步骤2.1.4:将相关系数按照从大到小排序,删除每个簇中前15%相关系数对应的节点;
步骤2.1.5:保留余下的节点作为新的簇
步骤2.1.6:令q=q+1,判断q是否大于k,如果是转到步骤2.2,否则转到步骤2.1.1;
步骤2.2:令更新后的簇集合
步骤2.2.1:利用改进l1正则化的特征选择算法,对输入的每个簇
步骤2.2.1.1:输入样本空间
步骤2.2.1.2:在样本空间x中随机采样
步骤2.2.1.3:用lasso回归模型计算损失函数e(x*,y*)+α‖w‖,其中w为惩罚项系数。
步骤2.2.1.4:将回归模型中xi对应的系数记为t,则证明该特征被选择,令其特征权重
步骤2.2.1.5:令i=i+1,判断i是否大于k,若是则转到步骤2.2.1.6,否则转到步骤2.2.1.2.
步骤2.2.1.6:输出所有xi对应的特征权重
步骤2.2.2:令w=w+1,判断w是否大于k,若是则执行步骤2.3,否则执行步骤2.2.1;
步骤2.3:计算每个特征的累加权重
步骤2.4:根据pw的排序结果,输出前l个特征作为最终的特征集合f={f1,f2,…,fl},其中f1对应pw中累加权重最大的一项;
步骤3:对于最终得到的特征集合f={f1,f2,…,f},从原始微阵列数据中找到对应的基因名称,完成对基因的特征分析。
本实施例在8种公开微阵列数据集上利用不同分类器分别进行了测试,如下表所示,测试中聚类簇k=5,重复抽样次数k=100,惩罚项α取0.3,选取的特征数目为10.
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明权利要求所限定的范围。
- 还没有人留言评论。精彩留言会获得点赞!