一种FASTQ文件的拆分方法、系统、电子设备及存储介质与流程
本发明涉及生物信息学,尤其涉及一种fastq文件的拆分方法、系统、电子设备及存储介质。
背景技术:
1、fastq格式是一种保存生物序列(通常为核酸序列)及其测序质量得分信息的文本格式。序列与质量得分皆由单个ascii字符表示。
2、该格式最初由维尔康姆基金会桑格研究所(wellcome trust sangerinstitute)开发,旨在将fasta格式序列及其质量数据整合在一起。而目前,fastq格式已经成为了保存高通量测序结果的事实标准。
3、fastq文件中,一个序列通常由四行组成:
4、第一行以@开头,之后为序列的标识符以及描述信息(与fasta格式的描述行类似)。第二行为序列信息。第三行以+开头,之后可以再次加上序列的标识及描述信息(可选)。第四行为质量得分信息,与第二行的序列相对应,长度必须与第二行相同。
5、以下为一个包含单个序列的fastq文件示例:
6、
7、其中!为最低质量、~则为最高质量。以下字符从左到右代表从低到高的质量得分的:
8、
9、通常,对于pe(paired-end,双端)测序,fastq文件为成对生成,se(single-end,单端)测序则生成单个fastq文件。
10、测序前的建库过程中存在把多个样品文库混合在一起的情况,通过加入barcode/index序列以区分不同的样品。barcode/index是为同一样品的每个原始dna连接上若干组(>=1)独一无二的序列编码,每组barcode通常由若干(>=1)段经过设计为核苷酸序列组成。根据设计不同,barcode出现在最终文件中的位置和方向不一而足,如图1(a)和图1(b)所示分别为常见的单索引和双索引的barcode/index序列的结构示例图。
11、而同批次建库测序的结果中,barcode出现在fastq文件中的位置和方向是一致的。测序完成后,根据已知的barcode/index序列和位置、方向信息拆分出每个样品的测序结果。
12、而同批次建库测序的结果中,barcode出现在fastq文件中的位置和方向是一致的。测序完成后,根据已知的barcode/index序列和位置、方向信息拆分出每个样品的测序结果。
13、现有技术中常见的根据barcode拆分fastq的软件,如seqtk_demultiplex、fastq-multx等,存在以下不足:
14、1、有些(如seqtk_demultiplex)只能支持single index(每组index只包含一个barcode片段)拆分;有些只支持single index或dual index(每组index只包含两个barcode片段)
15、2、当测序过程中双端序列反向不一致,需要进行二次拆分;
16、3、只能支持样品和barcode一一对映的场景;若一个样品存在多个(组)barcode,则需要分别拆分后再额外进行合并步骤;
17、4、只支持输入单个或成对fastq文件;
18、5、输出文件数量不可变(通常为成对fastq文件);输出序列长度和位置不可变。
技术实现思路
1、本发明针对现有技术中存在的技术问题,提供一种fastq文件的拆分方法、系统、电子设备及存储介质,解决现有技术中缺少一种通用的拆分方案的问题。
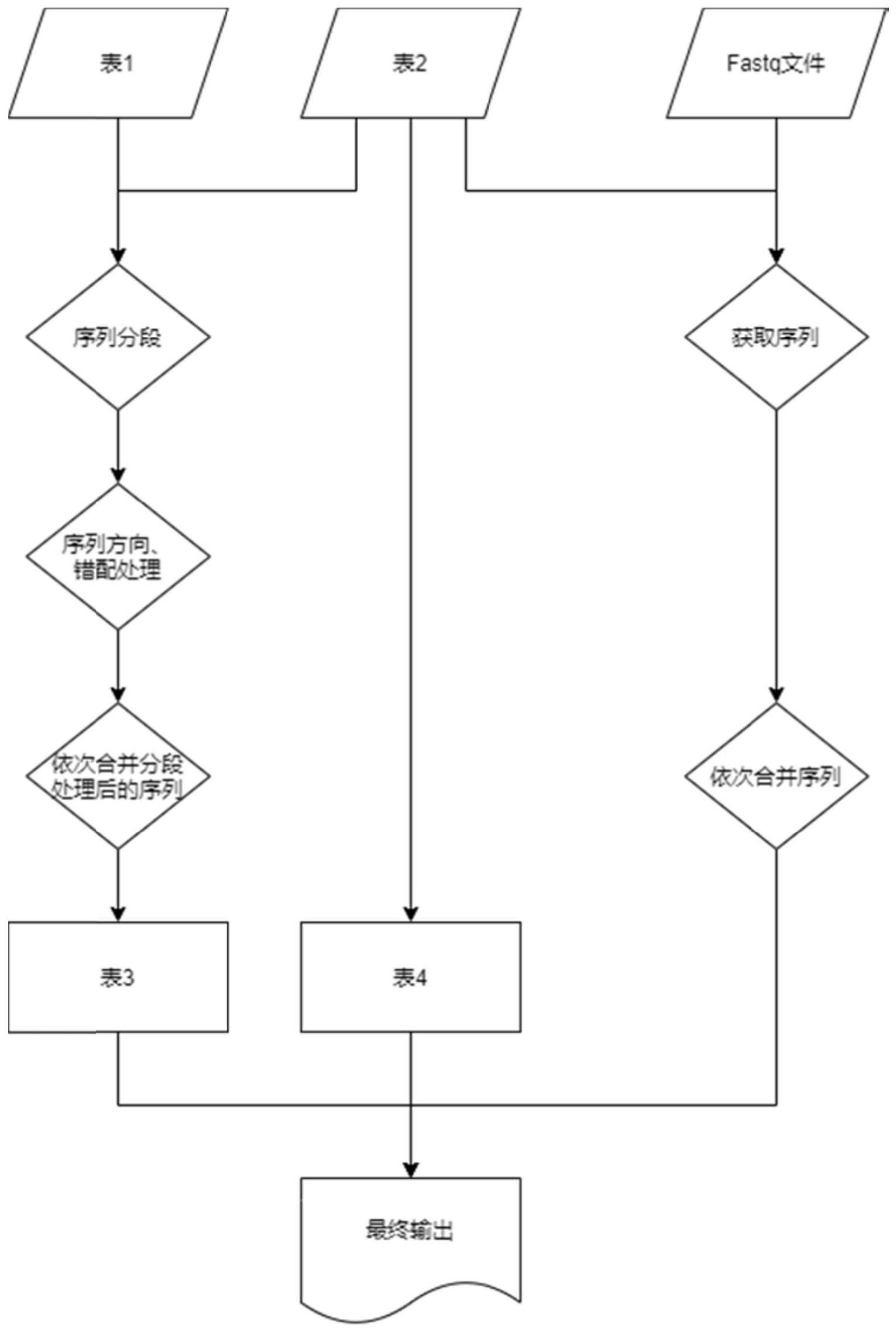
2、根据本发明的第一方面,提供了一种fastq文件的拆分方法,包括:以barcode/index序列为键、样品id为值构建表一;
3、以barcode/index序列为键、所述barcode/index序列所属fastq文件的序号及其在所述所属fastq文件中的位置信息为值构建表二;
4、对所述表一中的各组barcode/index序列进行分段、错配碱基替换以及合并处理后生成新的序列,以新的序列为键、样品id为值构建表三;所述表示连接顺序的字符串根据配错顺序以及所述表一的连接符生成;
5、构建表四包括:输出文件的序号、对应输入fastq文件序号、该文件输出序列的起始位置和长度;
6、以每4行为一条序列,同时遍历各个fastq文件,基于所述表一、表二、表三和表四对每个所述单位序列进行拆分。
7、在上述技术方案的基础上,本发明还可以作出如下改进。
8、可选的,所述表一、表二、表三和表四为哈希表。
9、可选的,构建所述表二和表四的过程中,所述barcode/index序列在其所属fastq文件中的位置信息包括:
10、起始位置、长度和方向,所述方向为正向或反向互补。
11、可选的,构建所述表三的过程中,对任意一组所述barcode/index序列的处理过程包括:
12、步骤1,根据所述表一的连接符,将所述barcode/index序列按顺序拆分成若干段序列并记为序列一;
13、步骤2,对各段序列一分别进行处理的过程包括:
14、步骤201,根据所述序列一的顺序序号和所述表二获取该序列一fastq在所属fastq文件中的位置信息,所述位置信息包括方向,所述方向为正向或反向互补;
15、步骤202,对所述序列一进行错配碱基替换处理,包括:
16、所述序列一的方向为反向互补时,将所述序列一做逆序处理,并对所述序列一的每个字符进行替换生成序列二,替换方式为:a变成t,t变成a,c变成g,g变成c;所述序列一的方向为正向时,所述序列二与序列一相同;
17、步骤203,生成与所述序列二有n个不一致字符的一组序列并记为序列三;n为所述步骤202中的错配数;
18、步骤3,依次从各组所述序列三中各取一条序列,将取出的各条序列根据所述表一中定义的连接符连接成字符串,以所述字符串为键、样品id为值生成所述表三。
19、可选的,所述对每个单位的数据进行拆分的过程包括:
20、遍历所述表二中的每个barcode/index序列的编号,根据所述表二的键值对应关系获取所述单位序列所属fastq文件的序号及其在所述所属fastq文件中的位置信息,获取所述所属fastq文件中位置信息对应的序列并记为序列四;
21、根据所述表二中的barcode/index序列的编号顺序,根据所述表一中的指定连接符将所述序列四连接为字符串并记为序列五;以所述序列五为键,基于所述表三获取样品id;
22、根据所述表四的所属fastq文件的序号及其在所述所属fastq文件中的位置信息,获取所述所属fastq文件中所述单位序列的位置信息对应的序列并记为序列六,基于所述表四中的输出文件的序号输出所述序列六和对应的样品id。
23、可选的,输出所述序列六和对应的样品id的过程包括:
24、若已存在包含以所述样品id为前缀的输出文件,则将所述序列六追加输出到该输出文件中;
25、否则,生成一个以所述样品id和对应的输出文件的序号为前缀的输出文件,并将序列六输出到所述输出文件中。
26、可选的,在遍历各个fastq文件的过程中,记录每个所述序列五出现的次数并作为最终统计结果输出。
27、根据本发明的第二方面,提供一种fastq文件的拆分系统,包括:表一构建单元、表二构建单元、表三构建单元、表四构建单元和拆分单元;
28、所述表一构建单元,用于以barcode/index序列为键、样品id为值构建表一;
29、所述表二构建单元,用于以barcode/index序列为键、所述barcode/index序列所属fastq文件的序号及其在所述所属fastq文件中的位置信息为值构建表二;
30、所述表三构建单元,用于对所述表一中的各组barcode/index序列进行分段、错配以及合并处理后生成新的序列,以新的序列为键、样品id为值构建表三;所述表示连接顺序的字符串根据配错顺序以及所述表一的连接符生成;
31、所述表四构建单元用于构建表四,所述表四包括:输出文件的序号以及所述表二;
32、所述拆分单元,用于以每4行为一个单位序列,同时遍历各个fastq文件,基于所述表一、表二、表三和表四对每个所述单位序列进行拆分。
33、根据本发明的第三方面,提供了一种电子设备,包括存储器、处理器,所述处理器用于执行存储器中存储的计算机管理类程序时实现fastq文件的拆分方法的步骤。
34、根据本发明的第四方面,提供了一种计算机可读存储介质,其上存储有计算机管理类程序,所述计算机管理类程序被处理器执行时实现fastq文件的拆分方法的步骤。
35、本发明提供的一种fastq文件的拆分方法、系统、电子设备及存储介质,通过表一定义每个样品对于的barcode,支持每个样品和任意数量barcode的映射关系。
36、通过表二定义拆分中需要用到的数据源编号、位置信息和方向信息,支持:
37、输入任意数量的fastq文件,包括罕见的3个以上fastq的场景;
38、每组barcode包含任意数量的序列,包括3段以上的bacode的组合;
39、对每段barcode指定位置和方向信息;
40、输出任意数量的fastq文件;
41、每个输出文件可以根据要求指定输出序列的长度与位置;
42、一次性生成最终结果,无需拆分后的合并和二次拆分操作;且有完整的统计结果。
- 还没有人留言评论。精彩留言会获得点赞!