癌凝集素分类预测方法及系统
本发明属于生物信息处理,具体涉及癌凝集素分类预测方法及系统。
背景技术:
1、癌凝集素是一类与癌症发展和转移密切相关的蛋白质。它们主要通过促进肿瘤细胞和血管内皮细胞之间的黏附作用来影响肿瘤的血管生成和转移过程。癌凝集素在肿瘤血管生成中起着重要作用,它能与内皮细胞发生相互作用,促进血管生成的过程,并且用于肿瘤细胞血管内壁进血液循环,实现转移和扩散。
2、研究表明,癌凝集素与肿瘤的血管生成和转移过程息息相关,在此抑制它的活性可能有于阻断肿瘤的血管生成和转移,为癌症治疗提供新的方向。目前,针对癌凝集素的研究仍在不断入和积极进行中。科学家们希望进一步了解癌凝集素的作用机制和调控途径,探索针对癌凝集素的治疗策略和药物研发以期为癌症患者提供更有效的治疗。癌凝集素在肿瘤的形成和转移过程中发挥着要作用,相关研究对于深入理解肿瘤生物学特性并发展更有效的癌症治疗手段具有重要意义。
3、对于传统的机器学习分类方法有支持向量机(svm)、随机森林(random forest)、逻辑回归(logistic regression)等。这些模型可以通过训练样本集对特征进行学习和训练,并在未知样本上进行分类预测。对于传统的机器学习在速度上都是有一定的优势,但是对于癌凝集素识别的准确率有着很明显的不足,并且识别的环境会被限制,抗噪声能力差,导致传统的特征分类预测一直都有着效率不高的问题。
4、因此,只有设计一种在各种环境都有抗噪能力并且识别准确率高、速度快的方法,才能真正解决癌凝集素分类预测问题。
技术实现思路
1、本发明是为了克服现有技术中,传统的机器学习分类方法在癌凝集素识别方面存在识别准确率低、抗噪声能力差以及容易受环境限制的问题,提供了一种在各种环境都有抗噪能力并且识别准确率高、速度快的癌凝集素分类预测方法及系统。
2、为了达到上述发明目的,本发明采用以下技术方案:
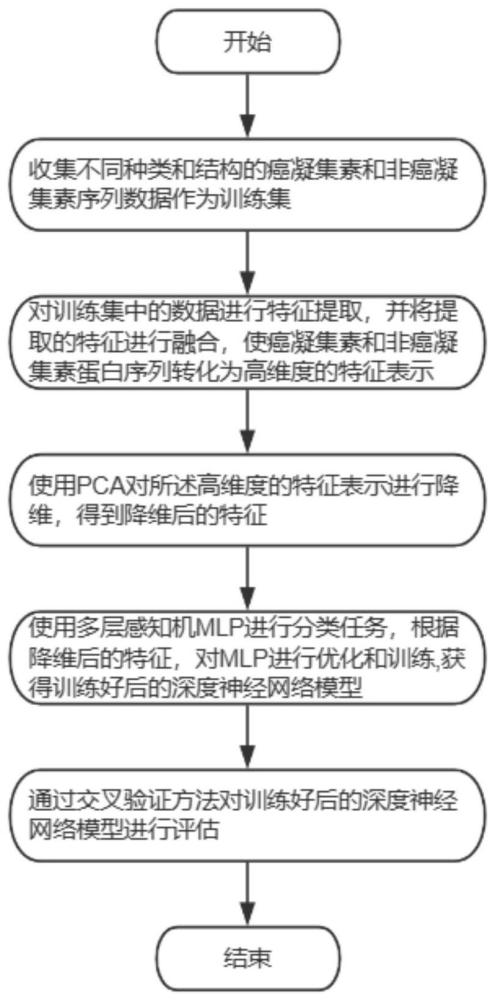
3、癌凝集素分类预测方法,包括如下步骤;
4、s1,收集不同种类和结构的癌凝集素序列和非癌凝集素序列数据作为训练集;
5、s2,对训练集中的数据进行特征提取,并将提取的特征进行融合,使癌凝集素和非癌凝集素蛋白序列转化为高维度的特征表示;
6、s3,使用pca对所述高维度的特征表示进行降维,得到降维后的特征;
7、s4,使用多层感知机mlp进行分类任务,并根据降维后的特征,对多层感知机mlp进行优化和训练,获得训练好后的深度神经网络模型;
8、s5,通过交叉验证方法对训练好后的深度神经网络模型进行评估。
9、作为优选,步骤s1包括如下步骤:
10、s11,从cancerlectiondb数据库中获取癌凝集素序列,从uniprot数据库中获取非癌凝集素序列,并使用cd-hit程序进行预处理,用于消除重复序列。
11、作为优选,步骤s2包括如下步骤:
12、s21,利用联合三元组描述符方法ctriad对癌凝集素与非癌凝集素进行特征提取,获得用于表示癌凝集素与非癌凝集素序列的特征;
13、s22,利用伪氨基酸组成方法paac对癌凝集素与非癌凝集素进行特征提取,获得用于表示癌凝集素与非癌凝集素序列的特征;
14、s23,利用k-间隔氨基酸对组成方法cksaap对癌凝集素与非癌凝集素进行特征提取,获得用于表示癌凝集素与非癌凝集素序列的特征;
15、s24,通过使用python编程将步骤s21、步骤s22和步骤s23中提取的三种特征按照维度进行合并。
16、作为优选,步骤s21包括如下步骤:
17、s211,将任意三个连续的氨基酸视为一个单位,来考虑一个氨基酸以及邻近氨基酸的属性;将蛋白质序列用二进制空间(v,f)表示,其中v表示序列特征的向量空间,每个特征vi表示一种三元组类型;f是v对应的数向量,其中f的第i维值fi是蛋白质序列中出现的vi型的数量。
18、作为优选,步骤s22包括如下步骤:
19、s221,定义
20、其中,表示第i个氨基酸的原始疏水性值,i的取值范围为1到20,h1(i)为第i个氨基酸的标准疏水性值;
21、采用同样的计算公式,通过第i个氨基酸的原始亲水性值计算出第i个氨基酸的标准亲水性值h2(i);通过第i个氨基酸的原始侧链质量mo(i),计算出第i个氨基酸的标准侧链质量m(i);
22、设定
23、上式中θ(ri,rj)相关函数是三种氨基酸性质的平均值;三种氨基酸性质具体为标准疏水性值、标准亲水性值和标准侧链质量;其中h1(ri),h1(rj)表示氨基酸ri和rj的标准疏水性值;h2(ri),h2(rj)表示氨基酸ri和rj的标准亲水性值;m(ri),m(rj)表示氨基酸ri和rj的标准侧链质量;
24、对于一个氨基酸的性质,氨基酸ri和rj对应的相关性θ(ri,rj)定义为:
25、θ(ri,rj)=[h(ri)-h(rj)]2
26、其中,h(ri),h(rj)为标准化后的氨基酸ri和rj的氨基酸性质;
27、对于一组n个氨基酸的性质,对应的相关性θ(ri,rj)定义为:
28、
29、其中,hk(ri),hk(rj)是氨基酸ri和rj的氨基酸属性集合中的第k个属性;
30、s222,定义一组被称为序列顺序相关因子的描述子为:
31、
32、其中,θ1,θ2,θ3,...,θτ表示分别间隔为1,2,3,...,τ的两个氨基酸ri和ri+1,2,3,...,τ的相关性,τ是一个要选择的整数参数,τ<n,n为整个序列长度;设fi为蛋白质序列中氨基酸i的归一化出现频率;一组20+τ的描述符,称为一个蛋白质序列的伪氨基酸组成,则定义为:
33、
34、其中,fc,fr分别为氨基酸c和r的归一化出现频率;c和r取值为[1,20],表示20种氨基酸;θc-20和θj分别为c-20和j间隔的氨基酸相关性取值,w为序列顺序效应的加权因子,设置为w=0.05。
35、作为优选,步骤s23包括如下步骤:
36、s231,计算由任意k个残基分隔的氨基酸对的频率;k的默认最大值为5;当k=0,则有400个0间隔的残基对,定义一个特征向量为:
37、
38、其中,a,c,d,y表示氨基酸的种类,如naa为丙氨酸和丙氨酸,nac为丙氨酸和半胱氨酸,nad为丙氨酸和天冬氨酸,nyy络氨酸和络氨酸,ntotal表示整个氨基酸序列。每个描述符的值表示蛋白质或肽序列中相应的残基对的组成;若残基对aa在蛋白质中出现m次,则残基对aa的组成等于m除以蛋白质中0个间隔的残基对的总数;对于k=0、1、2、3、4和5,长度为p的蛋白的ntotal值分别为p-1、p-2、p-3、p-4、p-5和p-6。
39、作为优选,所述pca为主成分分析,通过线性变换将m维的特征映射到k维,其中k<m。
40、作为优选,在步骤s4中,多层感知机mlp包括一个或多个隐藏层;每个隐藏层均包含多个神经元;
41、在多层感知机mlp中,神经元按层连接,并且每个神经元都与上一层的所有神经元连接;每个连接都有一个权重,用于调整数据在神经元之间传递时的重要性,同时还包括一个偏差用于调整神经元的激活阈值;
42、mlp通过训练数据集不断调整神经元之间的权重和偏差,使得深度神经网络模型学习到输入特征与输出之间的映射关系。
43、作为优选,在步骤s5中,使用如下评价指标来评价的模型的性能,包括:
44、准确性acc、敏感性se、特异性sp、马修斯相关系数mcc;
45、sp和se用于分别评价阴性例子和阳性例子的预测因子的预测性能;acc和mcc用于测量所有样例上的预测因子的整体能力。
46、本发明还提供了癌凝集素分类预测系统包括:
47、数据获取模块,用于收集不同种类和结构的癌凝集素序列和非癌凝集素序列数据作为训练集;
48、特征提取及融合模块,用于对训练集中的数据进行特征提取,并将提取的特征进行融合,使癌凝集素和非癌凝集素蛋白序列转化为高维度的特征表示;
49、特征降维模块,用于使用pca对所述高维度的特征表示进行降维,得到降维后的特征;
50、模型训练模块,用于使用多层感知机mlp进行分类任务,并根据降维后的特征,对多层感知机mlp进行优化和训练,获得训练好后的深度神经网络模型;
51、模型评估模块,用于通过交叉验证方法对训练好后的深度神经网络模型进行评估。
52、本发明与现有技术相比,有益效果是:(1)本发明中采用的模型结合了蛋白质的序列结构、氨基酸序列、物理化学性质等多种特征信息,通过深度神经网络进行综合学习和分类,这样的特征选择能够较全面地描述蛋白质的特性,提高分类的准确性;(2)本发明设计的深度神经网络模型保持了模型的简洁性和可解释性,通过有效地控制模型复杂度,使其能够更好地学习和分类蛋白质;(3)本发明中的模型采用了多层感知机(mlp)深度学习技术,能够高效对提取的蛋白质特征信息并进行分类,这样的设计能够加速分类过程,提高分类效率;(4)相比于传统的分类技术,本发明使用计算机技术使得分类成本大大降低,节省了大量的时间和资源成本,为进一步的研究打下坚实的基础;(5)从提高有关癌凝集素研究的进展来说,本发明可以为识别癌凝集素提供技术支持,具有极大的现实意义和广阔的应用场景。
- 还没有人留言评论。精彩留言会获得点赞!